Areas of Study
PROTEIN FOLDING, STABILITY AND INTERACTIONS
Proteins are the molecular machinery of the cell and are used to perform nearly every cellular function. Mutations have the potential to affect folding, stability and binding to its partners (other proteins, ligands, DNA/RNA). Our lab uses computational tools and techniques to address research problems in this area.
- We are analyzing the importance of individual residues in protein folding kinetics and developing algorithms using protein structural data to understand the mechanism of protein folding. These studies are expected to provide significant insights into protein structure formation, key contact networks in 3D structures and algorithms for predicting the folding kinetics of proteins.
- We have developed a knowledge-based methodology to predict the changes in folding rates upon mutations formulated from amino and acid properties using multiple linear regression. We have also developed a method for predicting protein unfolding rates upon mutations in two-state proteins by combining amino acid properties and knowledge-based classification of mutants with multiple linear regression.
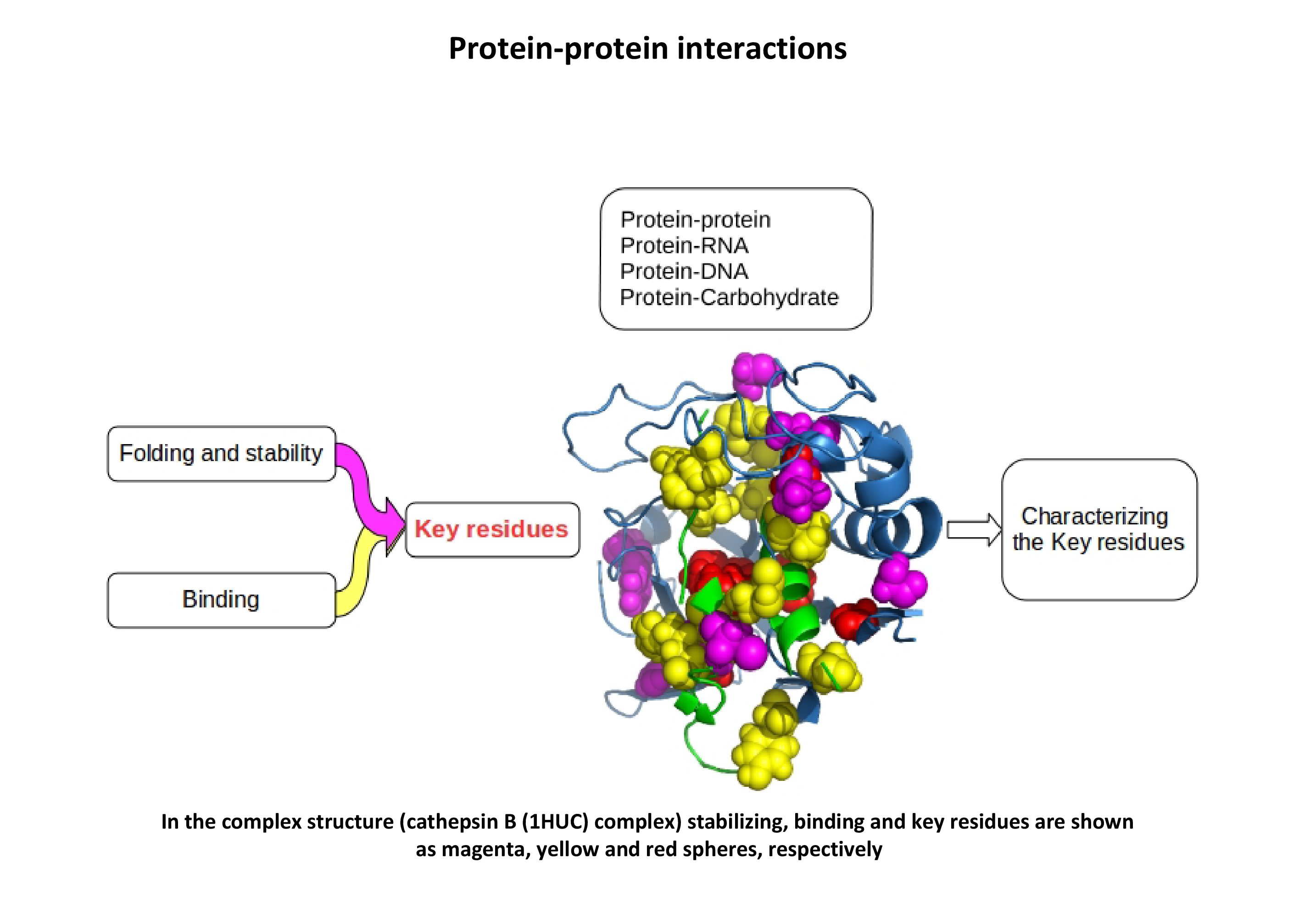
- We are working on residues which are involved in either binding or protein folding and stability, and key residues which are important for both processes. The key residues have been analyzed in various complex structures classified on the basis of protein structure, function, binding affinity and conformation. We are also investigating atomic contacts of binding and key residues, sequence and structural parameters such as surrounding hydrophobicity, solvent accessibility, secondary structure, long-range order as well as the conservation score of key residues.
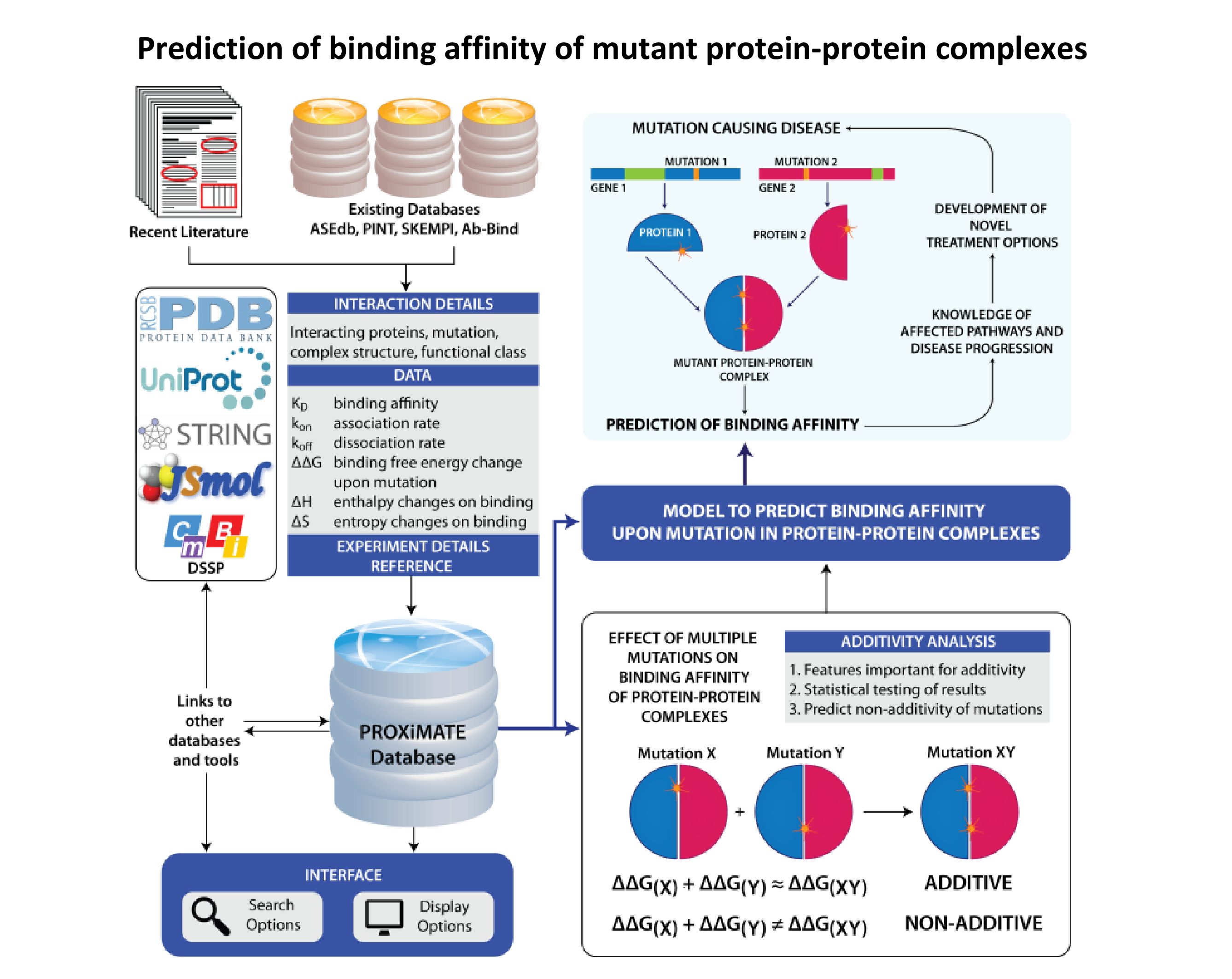
- We have collected data for the thermodynamics of mutant protein-protein interactions from literature and published it in a database, PROXiMATE (PROtein-protein CompleX MutAtion ThErmodynamics). We are analyzing double mutations in our database using structural features to distinguish between additive and non-additive data. We will use our analyses to build a model which can predict changes in binding affinity due to mutation in protein-protein complexes. The model can be used to improve our understanding of diseases and provide novel drug targets and therapy options.
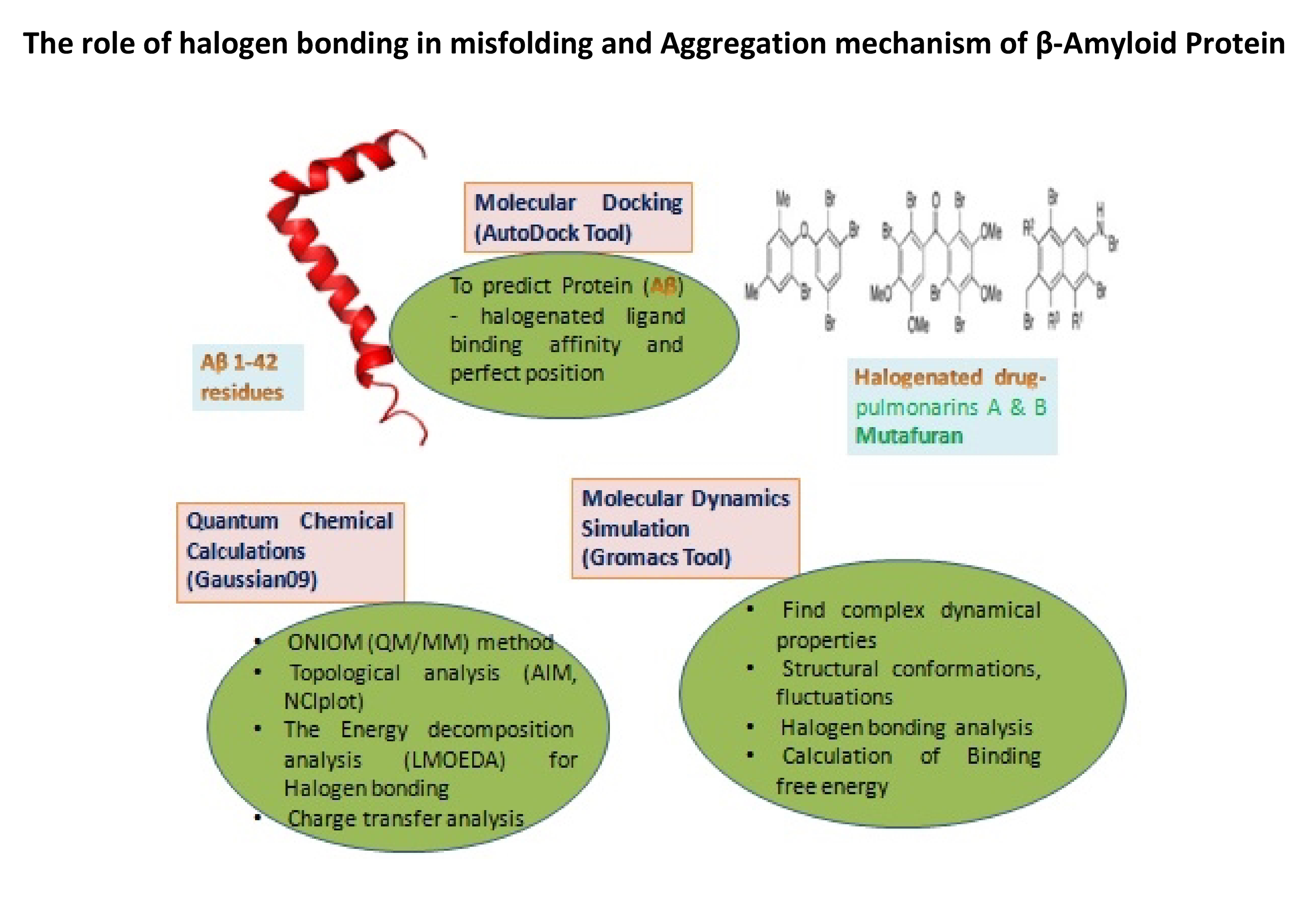
- We are also studying the interaction between halogenated drugs and amyloid beta proteins. We use molecular dynamics simulations to understand the binding affinity, free energy, structural conformation and other properties of mutafuran, pulmonarin A and pulmonarin B, with different substituted halogens. This study can be used to identify potential targets for halogenated drugs as a treatment for Alzheimer's disease.
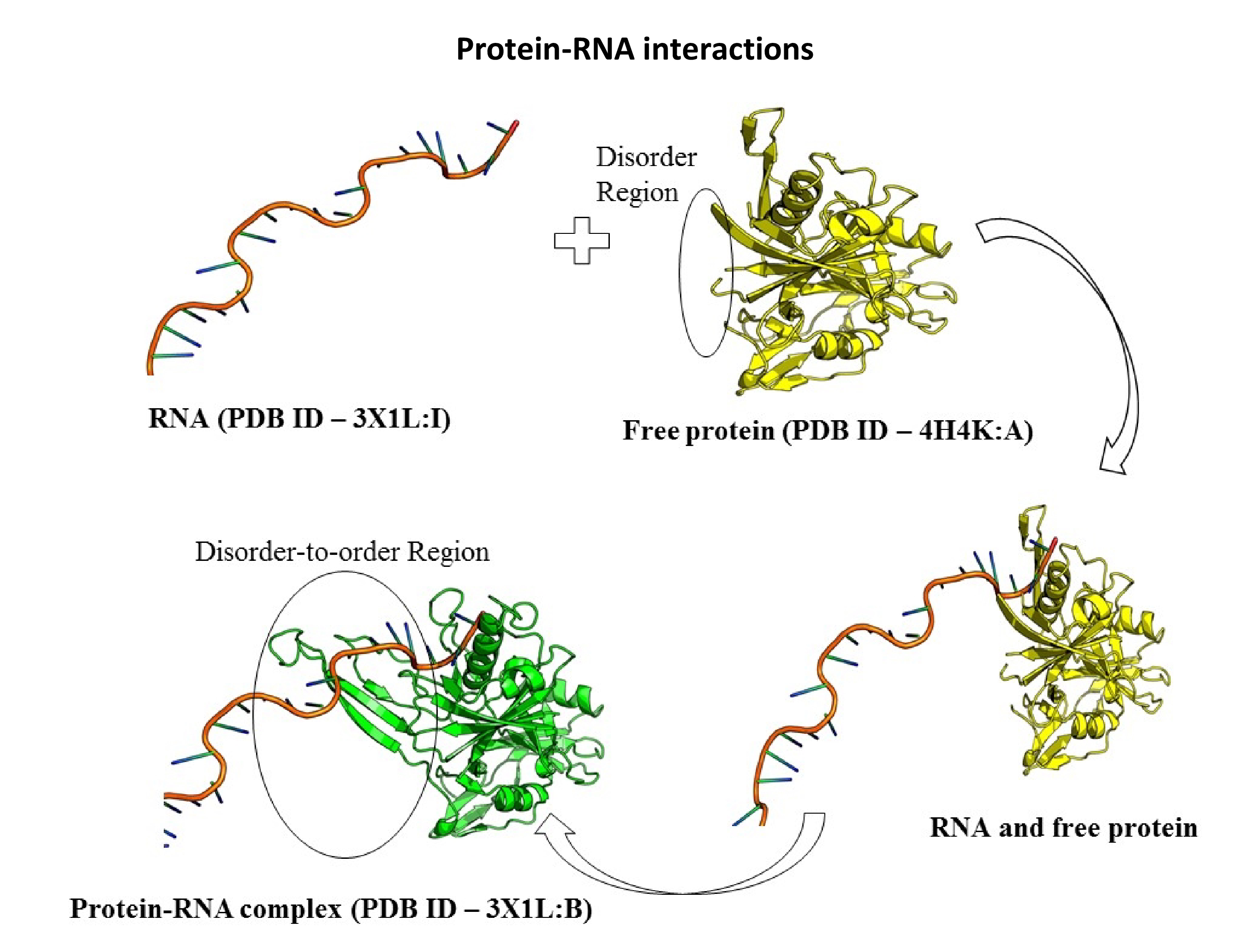
- We are working on understanding the interactions between disordered proteins and RNA. It is known that the disordered regions of proteins are capable transforming into ordered structures when interacting with other proteins, RNA, DNA or small molecules. Comparing available crystal structures for protein-RNA complexes and the unbound proteins helps us isolate the disorder-to-order transition (DOT) regions in the disordered proteins. Analyzing DOT regions for specific features has provided insights into the interactions of disordered proteins with nucleic acids.
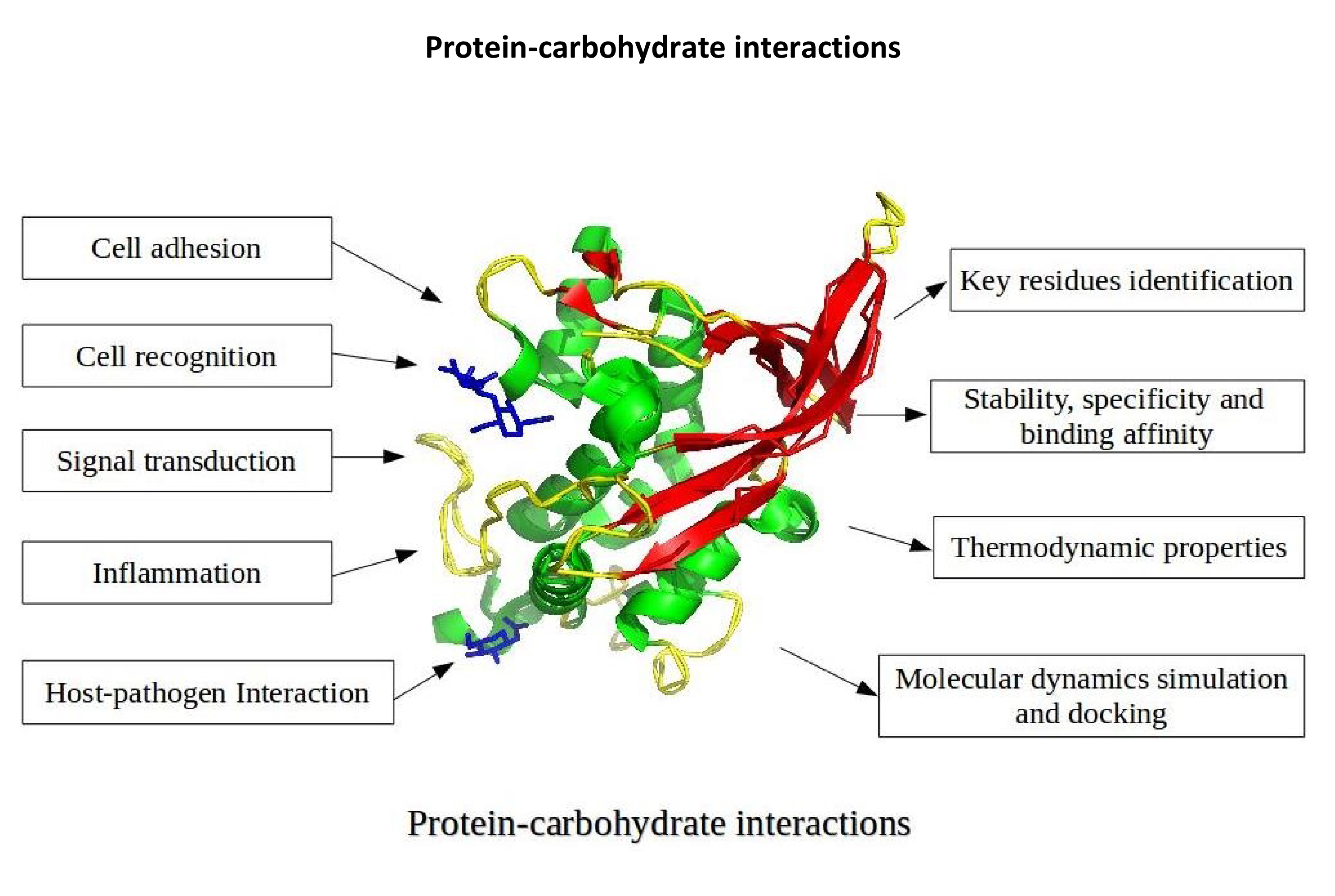
- Protein-carbohydrate interactions are essential for cell adhesion, recognition, signal transduction, host-pathogen recognition and inflammation. These interactions are often found at the cell surface, as part of a membrane glycoprotein. We have developed energy-based approaches for identifying binding sites and important residues for binding in protein-carbohydrate complexes. Lectins (proteins that interact with carbohydrates) are highly specific, the analysis of protein-carbohydrate interactions can be used to develop lectin-based carbohydrate biosensors.
References
- Jemimah, S., Yugandhar, K., and Gromiha, M. M. (2017). PROXiMATE: a database of mutant protein-protein complex thermodynamics and kinetics. Bioinformatics (in press).
- Gromiha, M. M., Yugandhar, K., and Jemimah, S. (2016) Protein-protein interactions: scoring schemes and binding affinity. Curr. Opin. Struct. Biol., 44: 31-38.
- Chaudhary, P., Naganathan, A. N. and Gromiha, M. M. (2015) Folding RaCe: A Robust Method for Predicting Changes in Protein Folding Rates upon Point Mutations. Bioinformatics, 31(13): 2091-7.
- Chaudhary, P., Naganathan, A. N. and Gromiha, M. M. (2016) Prediction of change in protein unfolding rates upon point mutations in two state proteins. Biochim. Biophys. Acta, Proteins Proteomics, 1864(9): 1104-9.
- Gromiha, M. M. and Yugandhar, K. (2017) Integrating computational methods and experimental data for understanding the recognition mechanism and binding affinity of protein-protein complexes. Prog. Biophys. Mol. Biol. (in press).
- Gromiha, M. M., Anoosha, P., and Huang, L.-T. (2016) Applications of Protein Thermodynamic Database for Understanding Protein Mutant Stability and Designing Stable Mutants. Methods Mol. Biol., 1415: 71-89.
- Magyar, C., Gromiha, M. M., Pujadas, G., Tusnády, G. E. and Simon I. (2005). SRide: a server for identifying stabilizing residues in proteins. Nucleic Acids Res., 33: W303-W305.
- Kulandaisamy, A., Lathi, V., ViswaPoorani, K., Yugandhar, K., and Gromiha, M. M., (2017). Important amino acid residues involved in folding and binding of protein-protein complexes. Int. J. Biol. Macromol., 94: 438-44.
PROTEIN AGGREGATION
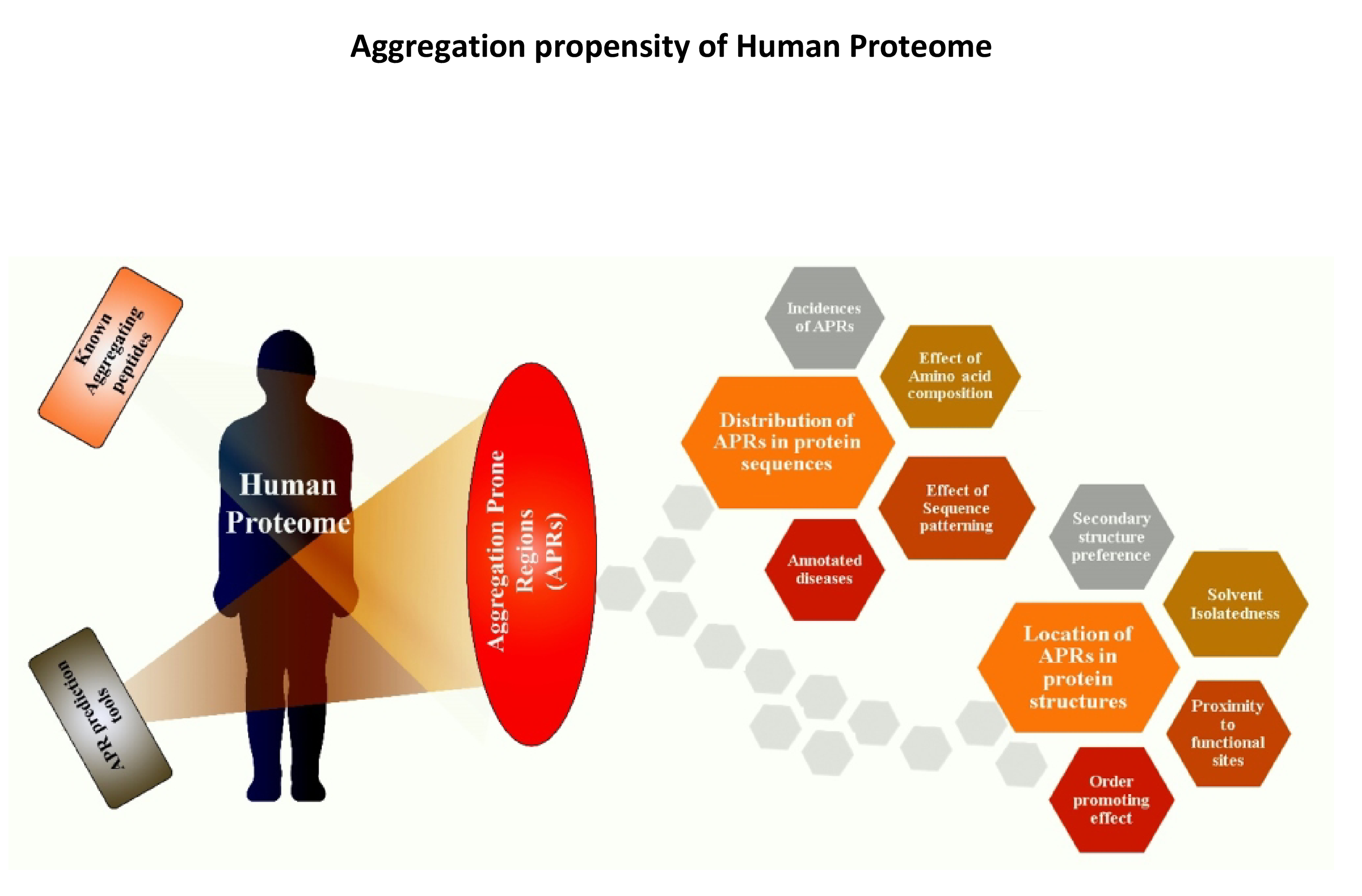
Protein aggregation has been implicated in many human disorders including Alzheimer's, Parkinson's, Huntington's, prion disease and Type II diabetes. Generally, a buildup of misfolded/unfolded proteins (i.e. a protein aggregate) is sequestered in inclusion bodies in the cell. This process may be disrupted for various reasons (such as mutations, stress and aging), resulting in the accumulation of protein aggregates within or outside the cell, leading to the formation of amyloids. These amyloids are associated with various disorders, particularly neurodegenerative diseases. Several in-vitro experiments have shown that even small environmental changes (pH, temperature, ionic concentration and additives) or substitutions in the protein sequence can have drastic effects on the rate of aggregation. Aggregation propensity depends on various factors, particularly the presence of aggregation prone regions (APRs) within the protein. These regions are capable of forming the cross-beta steric zipper motif which forms the stabilizing hydrophobic core of amyloid fibrils. We have developed a database and algorithms for the purpose of investigating protein aggregation.
- We have developed a Curated Protein Aggregation Database (CPAD), which contains results from experimental studies aimed at understanding protein/peptide aggregation. CPAD contains more than 3500 experimentally observed aggregation rates in native and mutated amyloidogenic proteins.
- We have also developed GAP (Generalized Aggregation Proneness), a tool to distinguish between amyloid fibril-forming and amorphous β-aggregating hexapeptides with almost 100% accuracy in non-redundant datasets. GAP can be applied in the design of novel biomaterials and improved aggregation inhibitors for biotechnological and therapeutic purposes.
- We have reported the presence of overlapping regions in the sequences of experimentally validated T-cell autoimmune epitopes, aggregating peptides, amyloidogenic proteins and randomly generated peptides. These regions drive aggregation and generate antibodies against soluble amyloid fibrils, and can be harnessed for early diagnosis of proteinopathies and the design of suitable drugs/vaccines against them.
- We are working on understanding APRs in human proteome and their association to human maladies. We have analyzed the occurrence of APRs in the entire human proteome using experimental data and predictions and we are using the results to refine GAP, our in-house APR prediction tool.
- We are also developing models to predict the aggregation rate. These models will help identify the underlying mechanism of aggregation and provide experimentalists with tools to design experiments in aggregation kinetics.

References
- Thangakani, A. M., Kumar, S., Nagarajan, R., Velmurugan, D. and Gromiha, M. M. (2014) GAP: Towards almost hundred percent prediction for β-strand mediated aggregating peptides with distinct morphologies. Bioinformatics, 30(14): 1983-90.
- Kumar, S., Thangakani, A. M., Nagarajan, R., Singh, S. K., Velmurugan, D. and Gromiha, M. M. (2016) Autoimmune Responses to Soluble Aggregates of Amyloidogenic Proteins Involved in Neurodegenerative Diseases: Overlapping Aggregation Prone and Autoimmunogenic regions. Sci. Rep., 6: 22258.
- Prabakaran, R., Goel, D., Kumar, S. and Gromiha, M. M. (2017) Aggregation Propensity of Human Proteome: Insights from large-scale data analyses. Proteins (in press).
- Thangakani, A. M., Nagarajan, R., Kumar, S., Sakthivel, R., Velmurugan, D. and Gromiha, M. M. (2016) CPAD, Curated Protein Aggregation Database: A Repository of Manually Curated Experimental Data on Protein and Peptide Aggregation. PLoS One, 11(4): e0152949.
MUTATIONS IN MEMBRANE PROTEINS AND DISEASE
Rapid progress in next-generation DNA sequencing technology has generated vast amounts of SNP (Single Nucleotide Polymorphism) data. SNPs occurring in gene-coding regions translate into missense mutations in proteins, with the potential to affect folding, stability, function and interaction. Mutations have been implicated in a number of human diseases and are of special interest in our lab. We use computational techniques to conduct large-scale analyses of mutations and also focus on mutations in proteins involved in specific diseases.
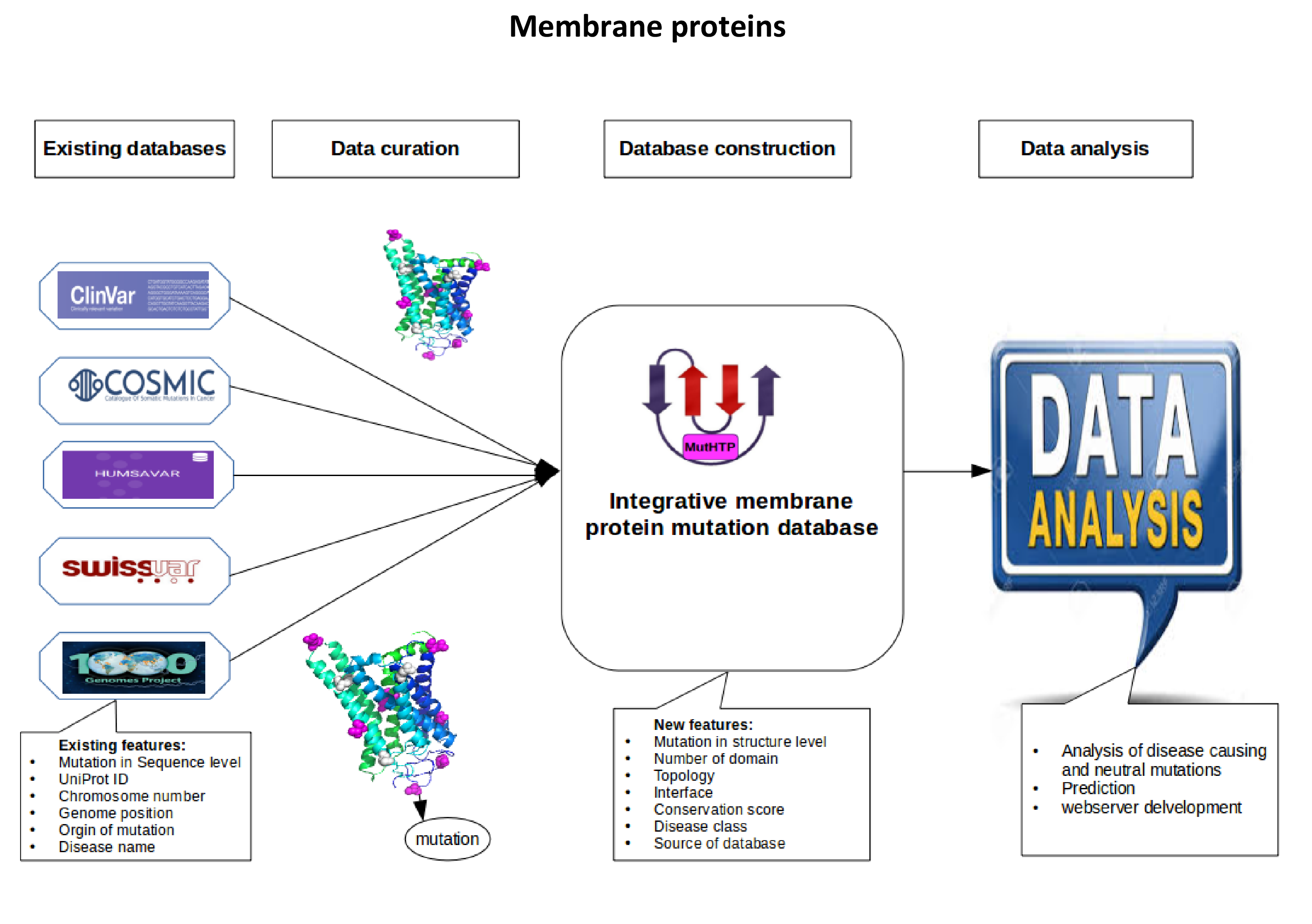
- We are working on the large-scale analysis of disease-causing and neutral mutations in human membrane proteins using computational approaches. We are developing an integrative database of disease mutations which will include comprehensive information, such as 3D structure, conservation score, topology, number of domains, interface and disease class of each mutation. Analyzing this data will help us build a prediction server to distinguish between neutral and disease-causing mutations.
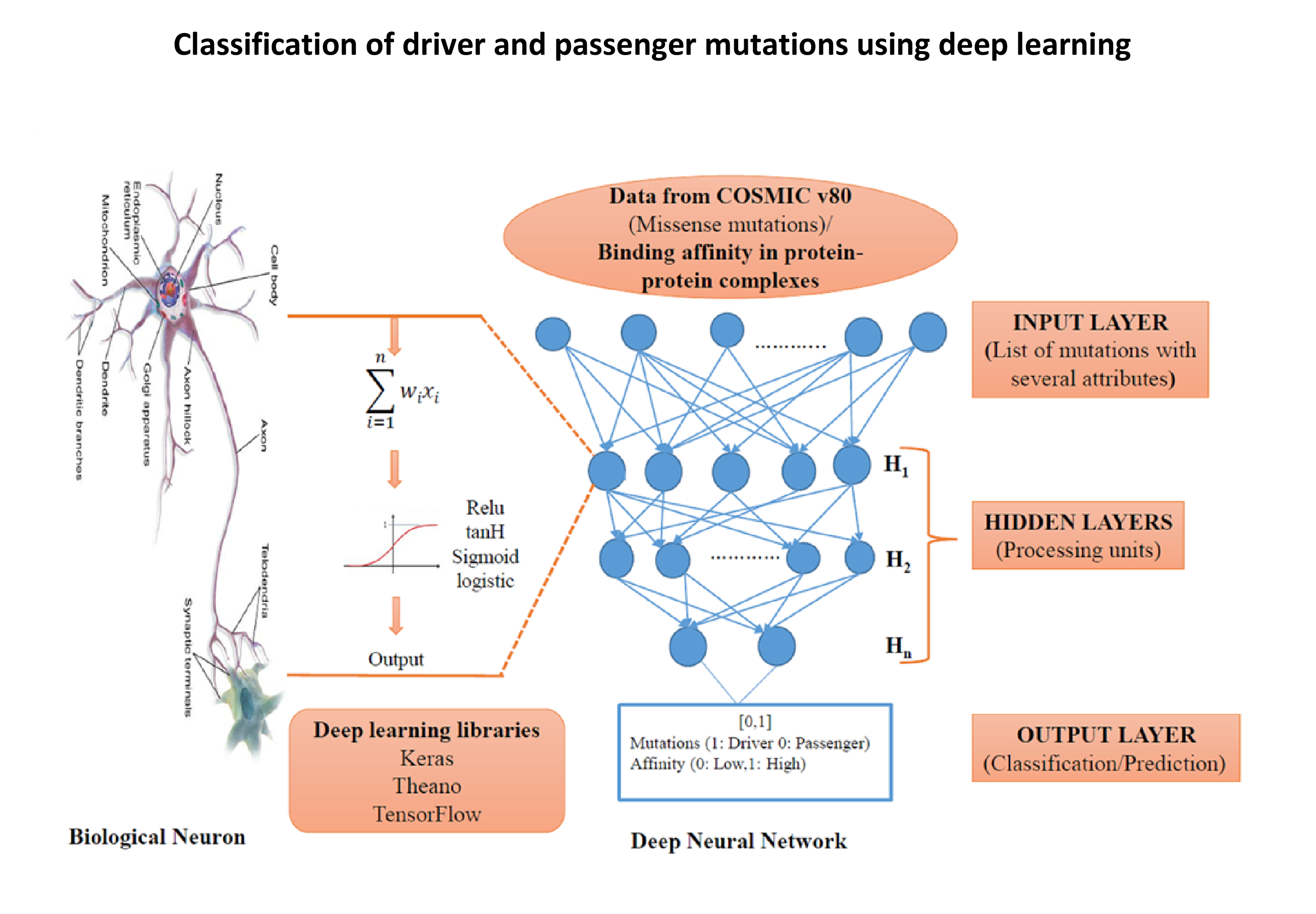
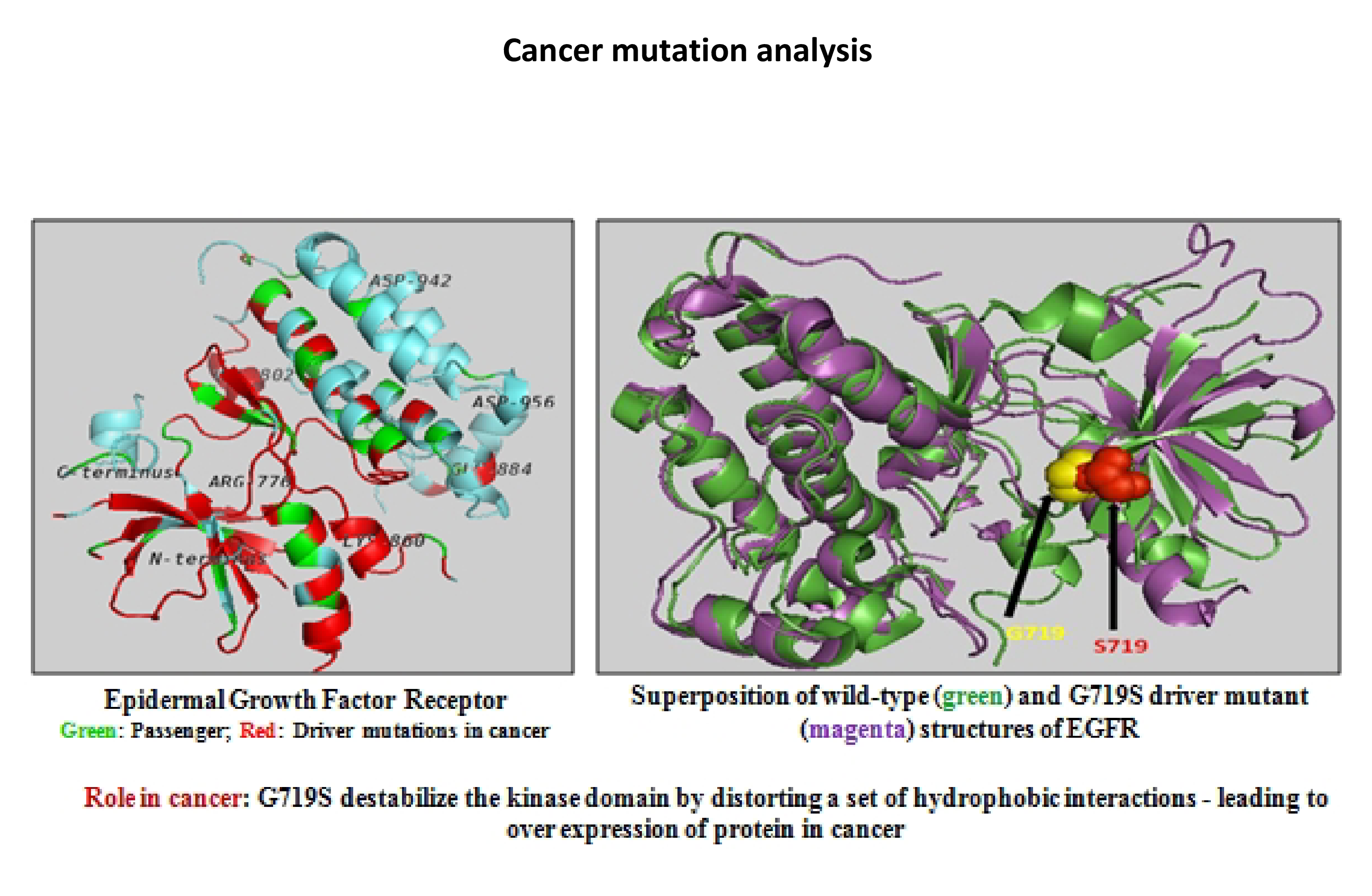
- We are also interested in discovering amino acid mutations relevant to cancer, a major challenge in the current cancer research. We mainly focus on proteome-wide analysis of cancer mutations to understand their role and explore mutational preferences in different cancer types. Our work includes the development of a robust binary classification model to discriminate between driver and passenger mutations in EGFR.
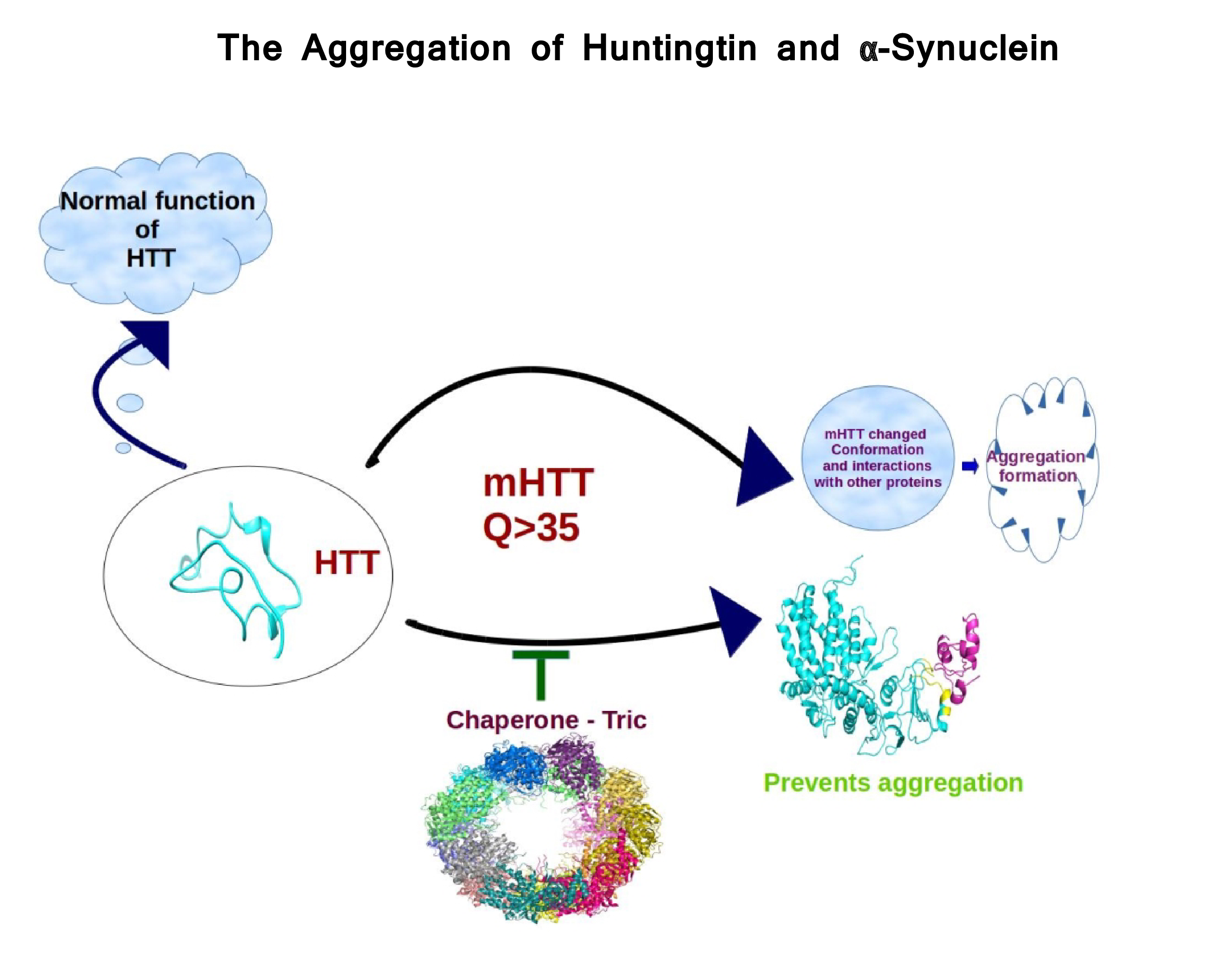
- We also focus on mutations involved in specific diseases. Huntington (HD) and Parkinson diseases (PD) are two major devastating neurodegenerative disorders of the central nervous system, associated with the accumulation of aggregate-prone proteins: mutant huntingtin (Htt) in HD and α-synuclein (α-syn) in PD. Although the functions of Htt and α-syn are unclear, mutations in Htt and α-syn promote aggregation. We use molecualr dynamics simulation and docking studies to study the the stability, interaction and aggregation behavior of the respective wild-type and mutant proteins. Understanding structural features and interactions can aid the development of novel strategies for therapeutic intervention.
References
- Anoosha, P., Sakthivel, R. and Gromiha, M. M. (2016) Exploring preferred amino acid mutations in cancer genes: Applications to identify potential drug targets. Biochim. Biophys. Acta, Mol. Basis Dis., 1862(2): 155-65.
- Anoosha, P., Huang, L.-T., Sakthivel, R., Karunagaran, D. and Gromiha, M. M. (2015) Discrimination of driver and passenger mutations in epidermal growth factor receptor in cancer. Mutat. Res. Fund. Mol. Mech. Mut., 780: 24-34.
- Michael Gromiha, M. M. and Ou Y-Y. (2014) Bioinformatics approaches for functional annotation of membrane proteins. Brief. Bioinf, 15(2), 155-168.
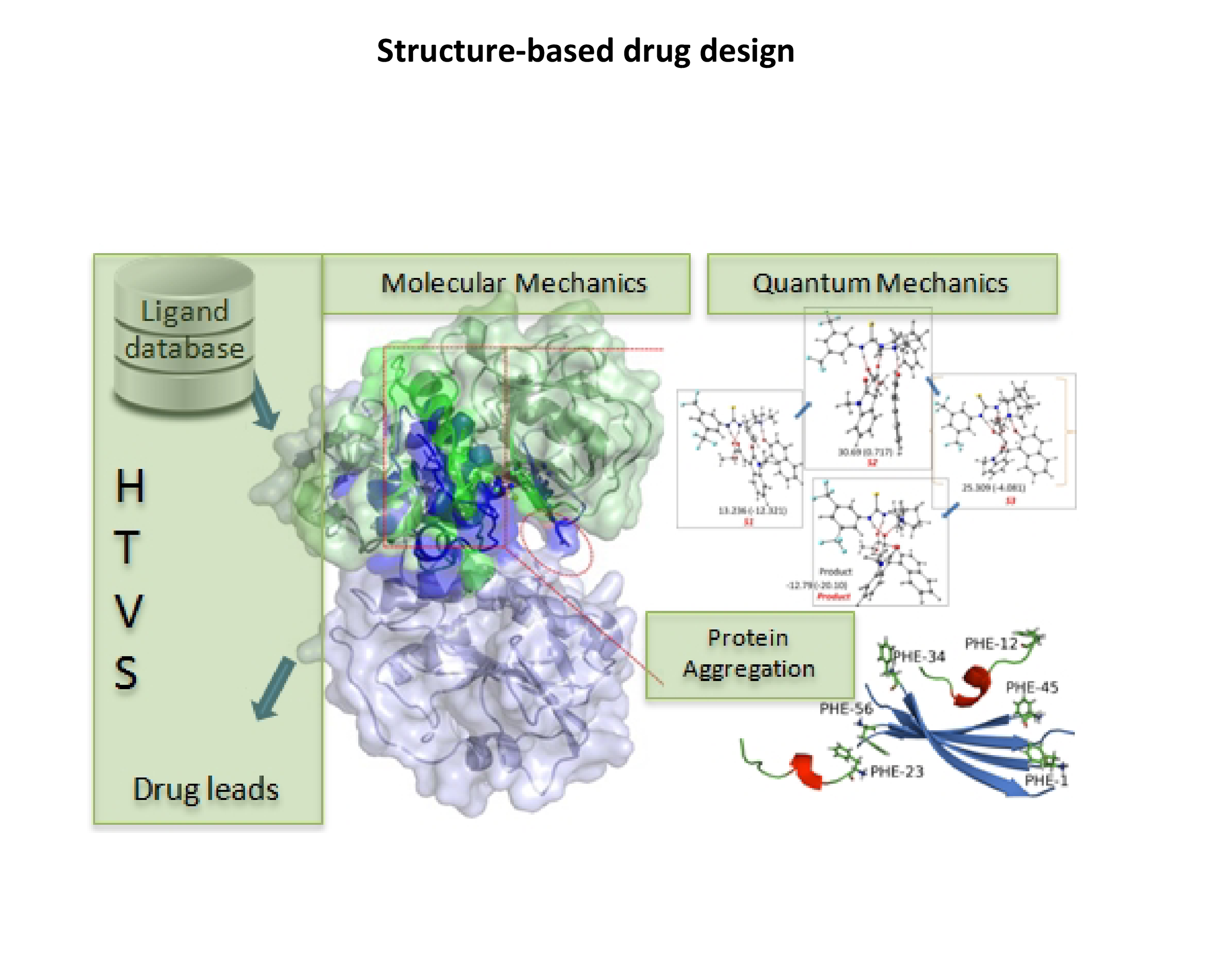
STRUCTURE-BASED DRUG DESIGN
Structure-based drug design is a major area of research in our lab.
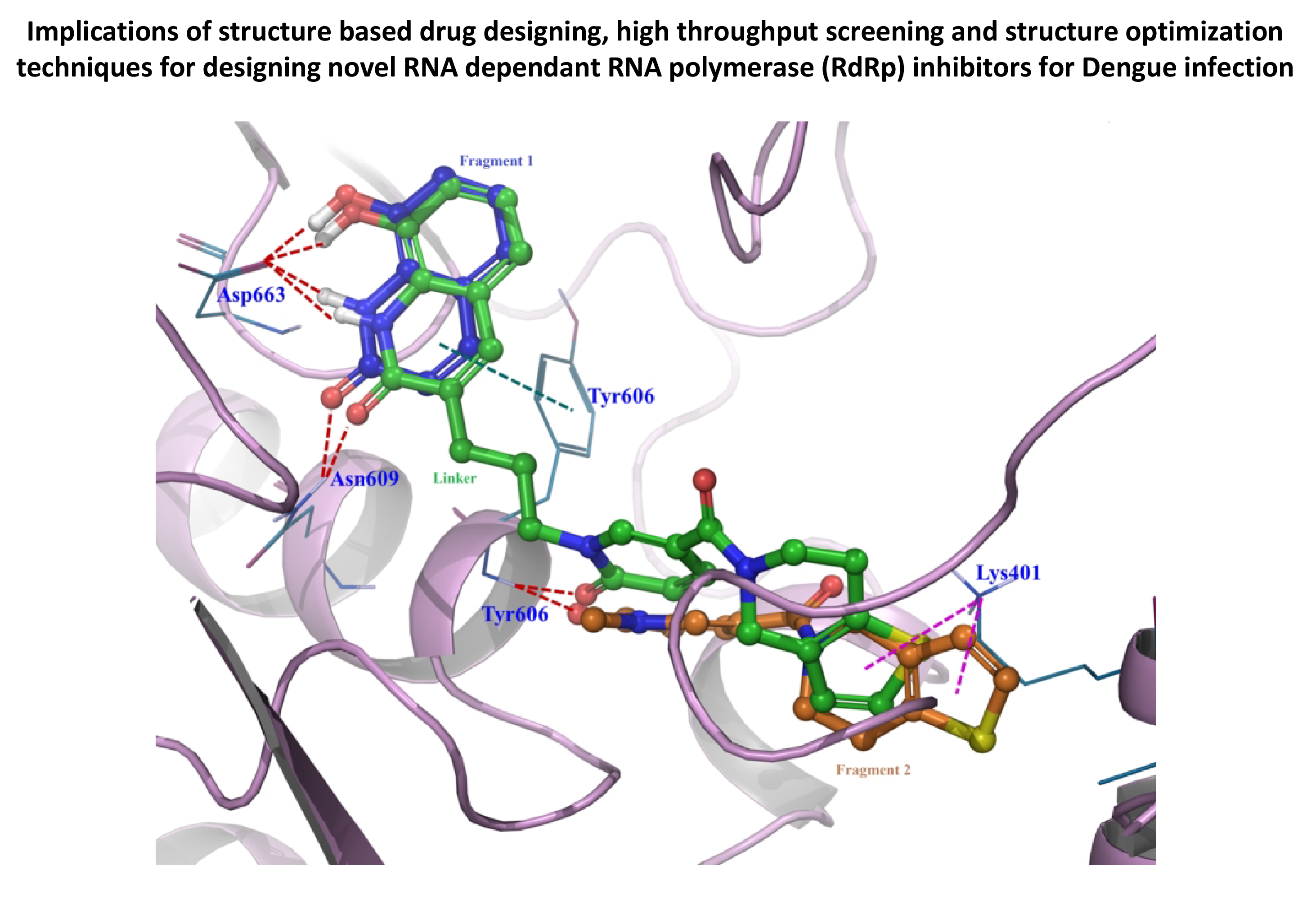
- We have identified a drug target and leads for dengue. Dengue is a vector-borne disease caused by the dengue virus (DENV). 50-100 million infections are reported every year but there are no specific therapeutics available to treat dengue. We chose RNA dependant RNA polymerase (RdRp, essential for dengue viral replication) as a drug target for designing a potent lead molecule for dengue. We used fragment-based approaches to design an inhibitor for RdRp, and validated the results using molecular dynamics simulation. QSAR was used for optimization. We also worked on flavanoids in our search for natural anti-dengue compounds.
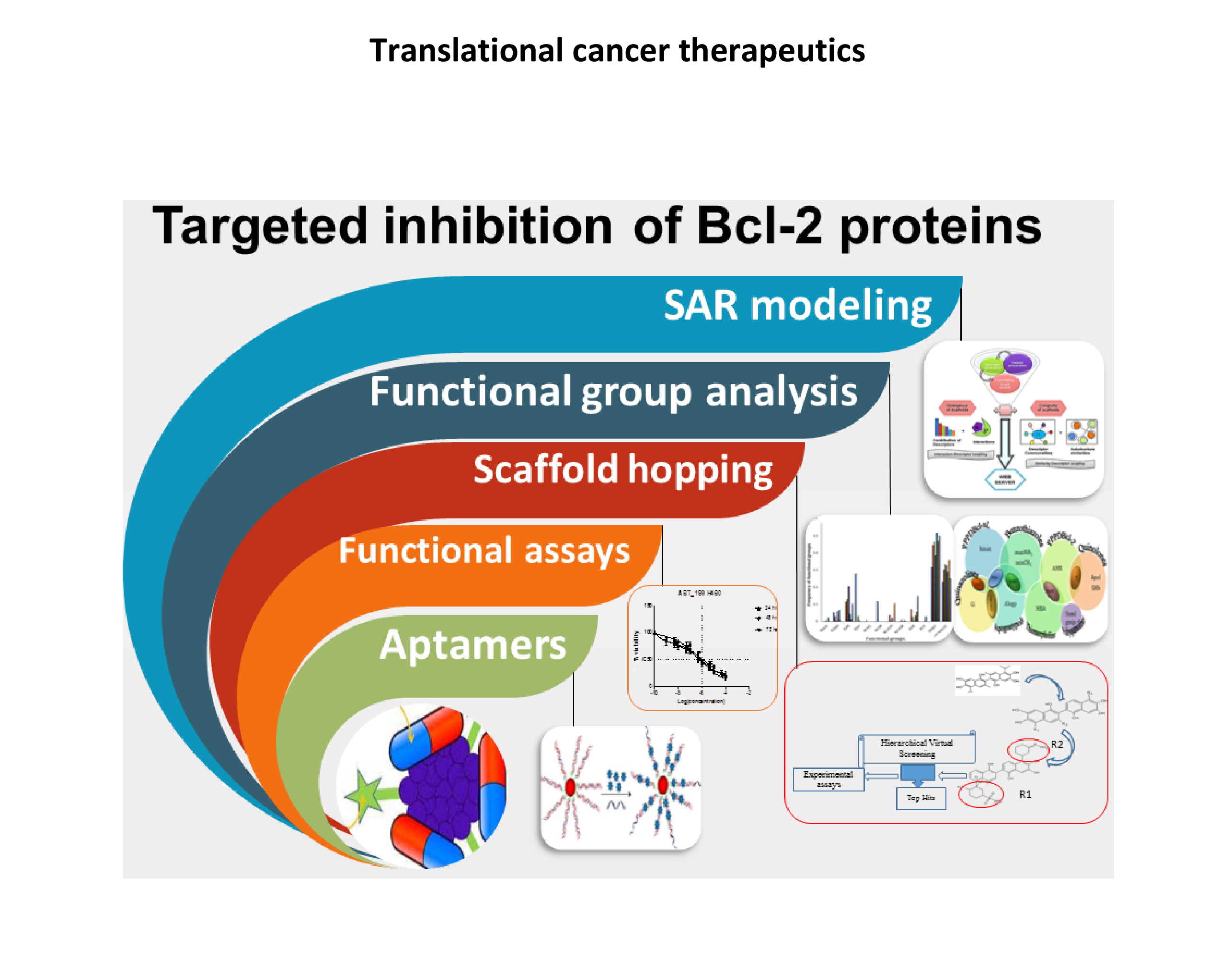
- We are developing novel therapeutics for cancer by targeting members of the Bcl-2 family using in-silico and in-vitro approaches. Our work includes the development of algorithms and web based tools for predicting novel inhibitors for Bcl-2 family proteins, deciphering the role of functional groups in reported inhibitors, designing of novel lead compounds by scaffold hopping and designing aptamer hybrids for targeting Bcl-2 proteins. We are carrying out experiments to confirm the efficacy of our novel compounds against cancer.
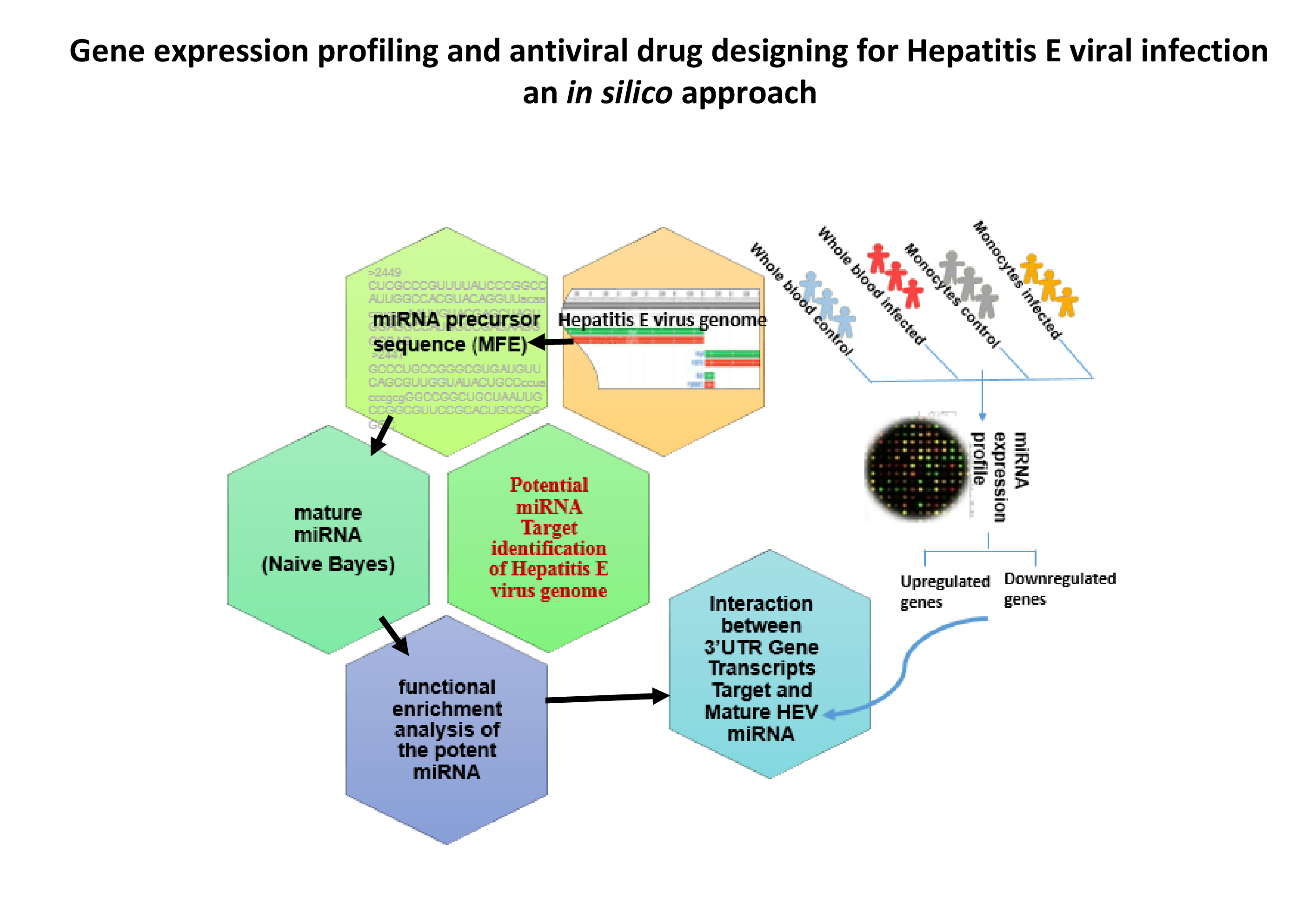
- We are also studying drug targets for Hepatitis E, a liver disease caused by Hepatitis E virus (HEV). Out of approximately 20 million infections worldwide, 3.3 million become symptomatic cases, leading to around 56600 deaths annually (Hepatitis E Factsheet from WHO, 2016). We are using computational approaches to understand HEV and identify potential drug targets in HEV using an RNA-based approach. We intend to identify potential drug targets using RNA secondary structure characteristics, genetic conservation and interaction features at seed regions. The outcome of this study can be applied for the development of drugs against HEV.
- We are working on the identification of inhibitors for c-Yes kinase and sirtuin using structure-based drug design and machine learning. We also use molecular dynamics simulation to study the the mechanism of aggregation of wild type and mutant keratoepithelin (KE). In collaboration, we are investigating the effect of organo-catalysts and enzymes in reaction mechanism using quantum mechanics modeling.
References
- Anusuya, S. and Gromiha, M. M. (2017) Quercetin derivatives as non-nucleoside inhibitors for dengue polymerase: molecular docking, molecular dynamics simulation and binding free energy calculation, J. Biomol. Struct. Dyn., DOI: 10.1080/07391102.2016.1234416.
- Anusuya, S., Velmurugan, D. and Gromiha, M. M. (2016) Identification of Dengue Viral RNA Dependent RNA Polymerase Inhibitor using Computational Fragment Based Approaches and Molecular Dynamics Study. J. Biomol. Struct. Dyn., 34(7): 1512-32.
- V. Kanakaveti, R. Sakthivel, Suresh K. Rayala and M. Michael Gromiha (2017) Importance of functional groups in predicting the activity of small molecule inhibitors of Bcl-2 and Bcl-xL. Chem. Biol. Drug Des. (in press).
- Ramakrishnan, C., Thangakani, A. M., Velmurugan, D., Krishnan, D. A., Sekijima, M., Akiyama, Y. and Gromiha, M. M. (2017) Identification of type I and type II inhibitors of c-Yes kinase using in silico and experimental techniques. J. Biomol. Struct. Dyn., 1-11.
- Chiba, S. et al. (2015) Identification of potential inhibitors based on compound proposal contest: Tyrosine-protein kinase Yes as a target. Sci. Rep., 5: 17209.
NEXT-GENERATION SEQUENCING AND DEEP LEARNING
NGS and deep learning represent significant advances in technology and will have a profound effect on research in computational biology.
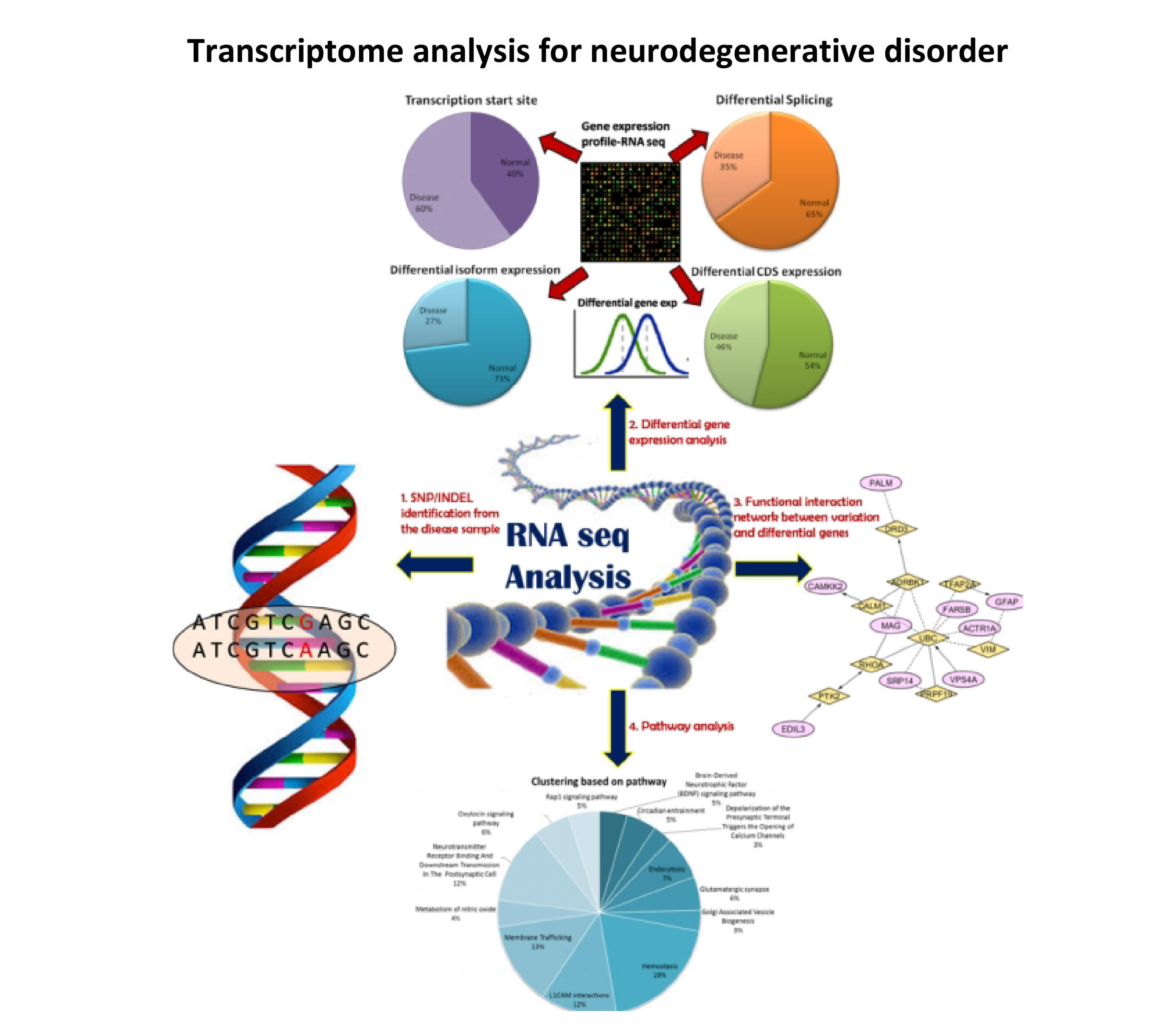
- An important aspect of neurodegenerative diseases like Alzheimer's and Parkinson's is neuronal death caused by protein aggregation, mitochondrial stress, aging and other factors. However, drugs used to treat this disease (Levo-dopa) do not improve neuronal survival. We work on transcriptome sequence data derived from next-generation sequencing technology to study gene expression profiles for affected individuals and identify biomarkers. We also analyze the crosstalk between pathways of neurodegenerative disorders. We are classifying suitable candidates for drug repositioning to devise treatment strategies which can rescue surviving neurons.
- Deep learning can be utilized to obtain insights from vast amounts of genomics and proteomics data. We have applied deep learning to the problem of classifying missense mutations in cancer, using missense mutations which are frequently found in various types of cancer. Our aim is to build a classification model using deep learning that can be applied to a large dataset of missense mutations.