Tutorial
Database of Experimental neutralization profile of coronavirus related antibodies
The Ab-CoV database contains information about experimental details related to neutralization and binding affinity of antibodies. The database can be searched based on followingn criteria:
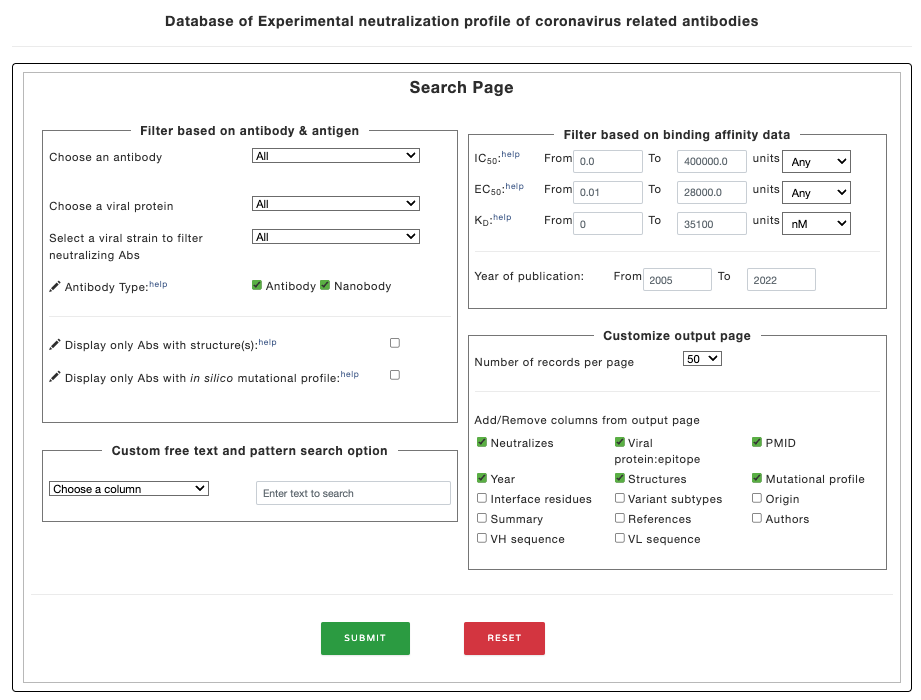
Figure 1. Filter options in the main page.
The search option leads to a table with following columns. Users have option to choose the columns to display
The details of the columns are given below:
| Column/Property | Description |
|---|---|
| Entry | Unique IDs are given to each antibody sequence |
| Antibody name | Antibody name |
| Type | Antibody or Nanobody |
| IC50 | Half maximal inhibitory concentration |
| EC50 | Half maximal effective concentration |
| Binding affinity | Strength of the binding interaction (KD) |
| Neutralizes | Coronavirus strain neutralized by antibody |
| Viral protein:epitope | Viral protein and domain associated with antibody binding |
| PMID | Pubmed ID |
| Year | Year of publication |
| Structures | PDB structure of the antibody |
| Mutational profile | Link to internal dataset of mutational scanning of epitope and paratopes (if experimental complex structure information is available) |
| Interface residues | The list of epitope and paratope residues (if mutational profile exists) |
| Variant subtypes | Variant of coronavirus for which antibdy is tested |
| Origin | Source organism of antibody |
| Summary | Short summary from the respective research paper |
| References | Reference for the publication |
| Authors | List of authors |
| VH sequences | Heavy chain sequence of antibody variable region |
| VL sequences | Light chain sequence of antibody variable region |
The output table for the dataset looks like below:
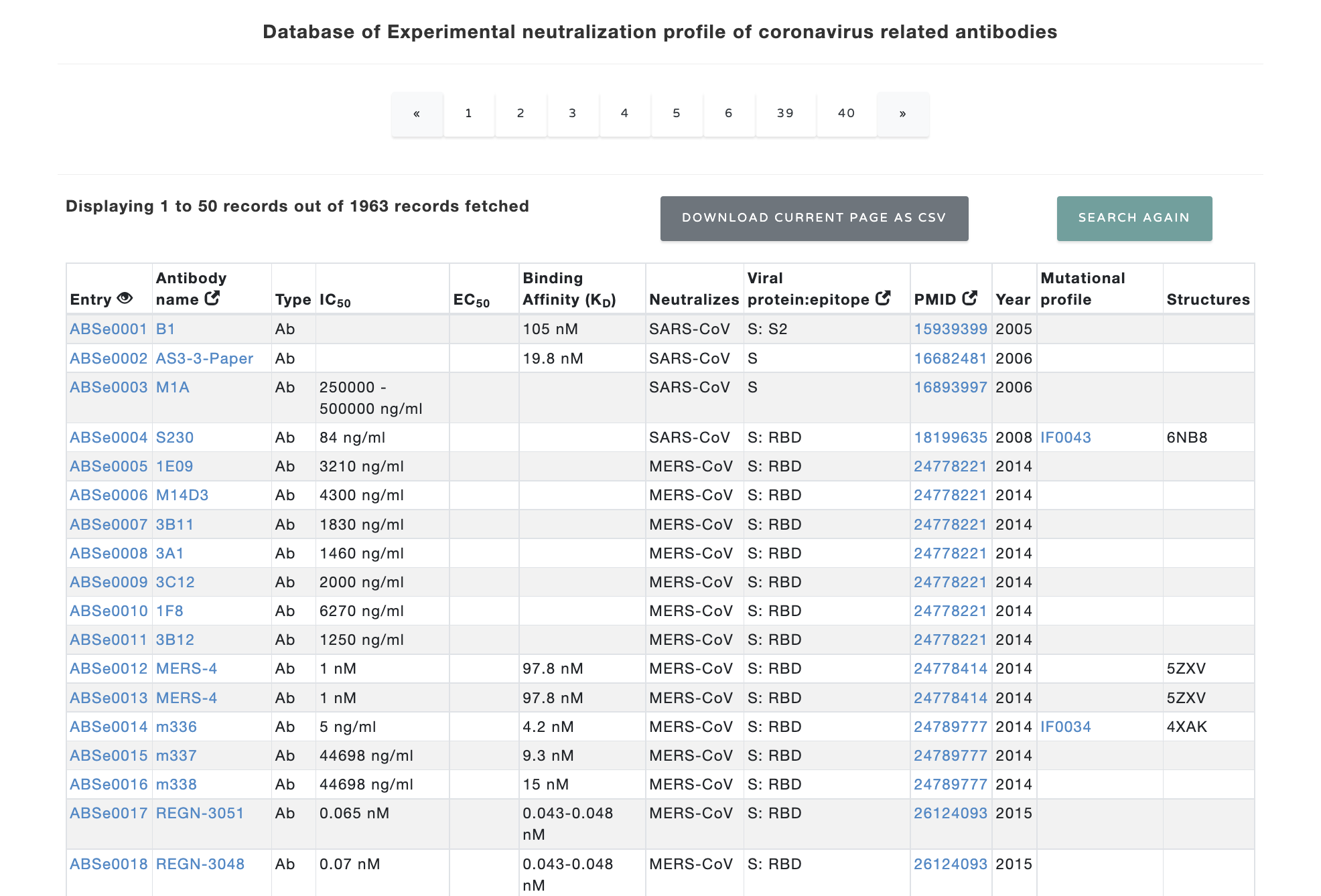
Figure 2. Table at the main page.
We have also collected/calculated some additional information for the antibody sequences. Each row in the result table is clickable and provides additional information about the neutralizing antibody. For each entry, the result page contains IMGT Collier de Perles graph for heavy chain and light chain, top 10 similar therapeutic monoclonal antibodies with their details, modelled structure, experimental detail (IC50, EC50 and KD values) and literature information. The "overall percentile" column shows the position of respective IC50/EC50/KD value among all IC50/EC50/KD values in the Ab-CoV database as percentile to rank the antibodies. The result page looks like as shown below:
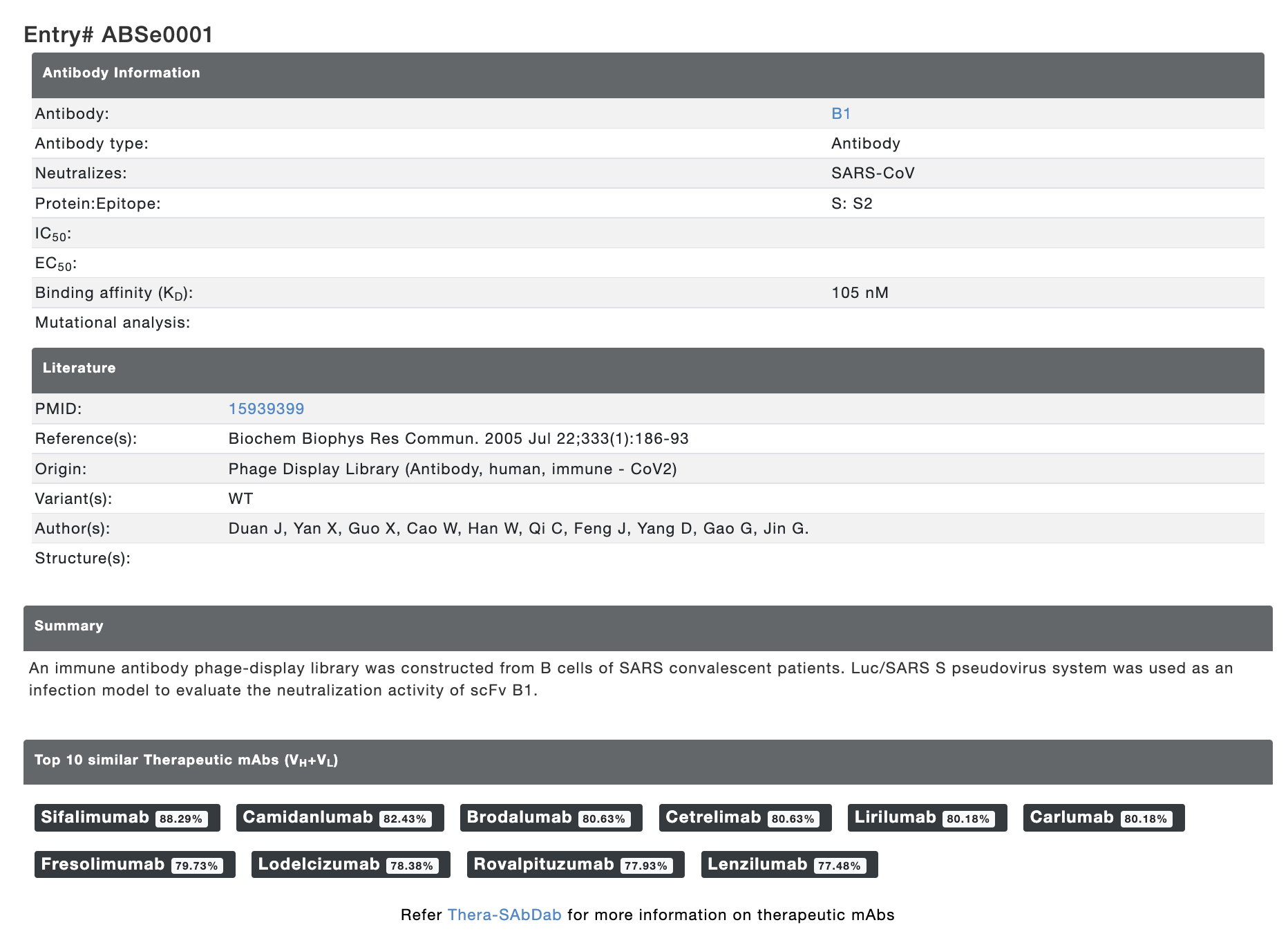
Figure 3. Additional calculated information and experimental details provided for each entry.

Figure 4. IMGT Collier de Perles graph for heavy chain and light chain provided for each entry.
The top 10 similar therapeutic antibodies are listed with % sequence identity (VH+VL sequence). The therapeutic antibodies are not part of the database. This resource can be accessed from Thera-SAbDab database. Similarly, the sequences of neutralizing antibodies can be accessed from CoV-AbDab database.
Additional information: dataset for in silico mutational scanning of the spike-antibody interface
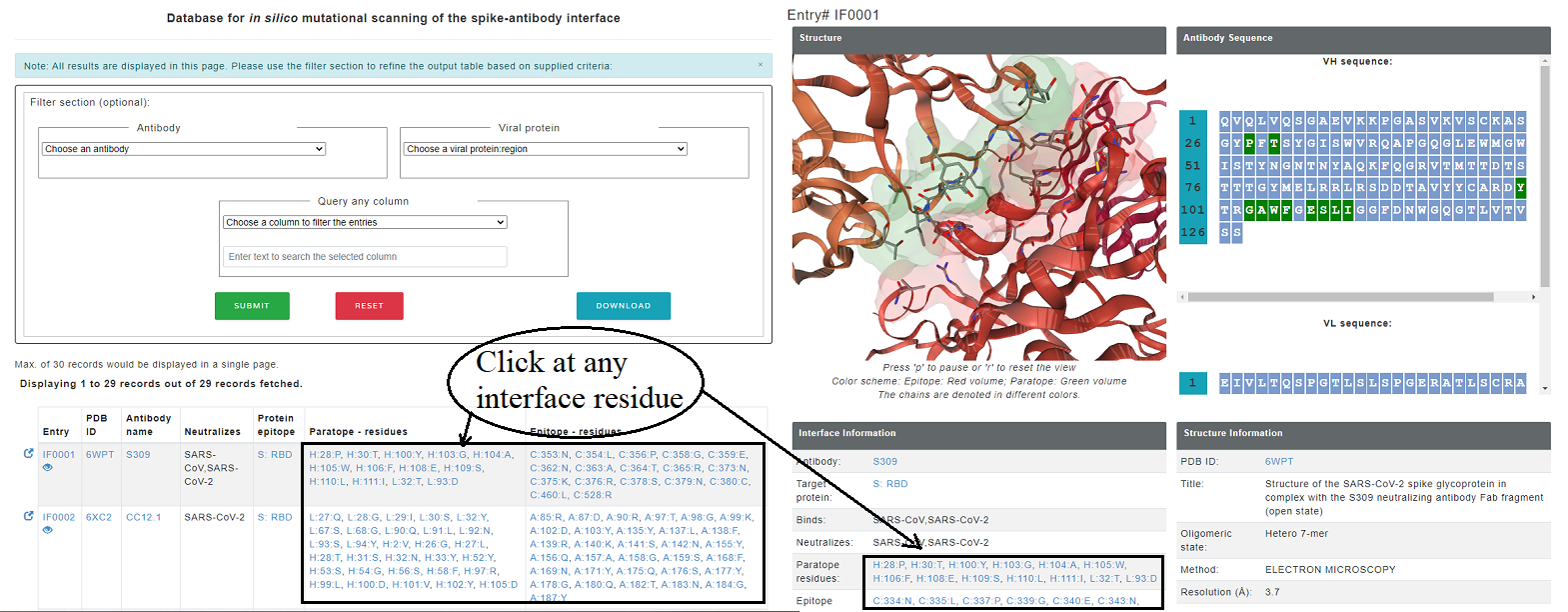
Some entries in Neutralizing antibody dataset also has a experimentally determined viral protein-antibody structure. For these antibody complexes, the effect of mutation on stability and binding affinity is assessed using computational tools including FoldX (stability and binding affinity, emperical function based), mCSM (stability and binding affinity, graph-based), CUPSAT (stability, amino acid-atom potentials and torsion angle distribution) and ProAffiMuSeq (binding affinity, machine learning based using sequence-based features). There are seperate tables for the epitope and paratope residues and each method gives affect on stability/binding affinity for 19 mutations. For each entry, the result page displays the structure of the antibody with the interface residues highlighted as sticks. interface residues are also highlighted in the sequence of antibody VH and VL chains in green color. Moreover, each entry for paratope and epitope residues are linked to comprehensive mutational information.
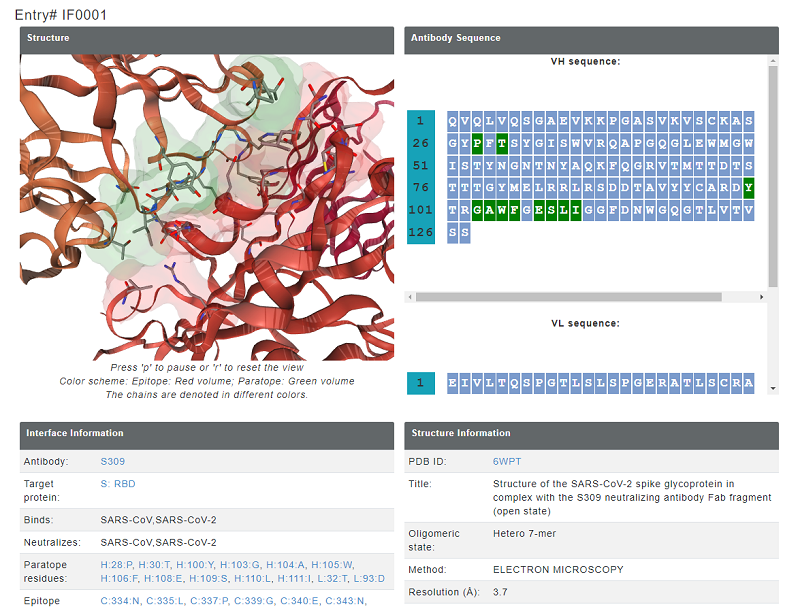
Figure 5. epitoep and paratope inforamtion provided for each entry. In the structure, paratope is shown in blue colored volume and epitope is shown in red colored volume.
The mutation data has the values for binding affinity change upon mutation and stability change upon mutation in kcal/mol from mCSM, ProAffiMuSeq, CUPSAT and FoldX. The images from the result page is shown below:
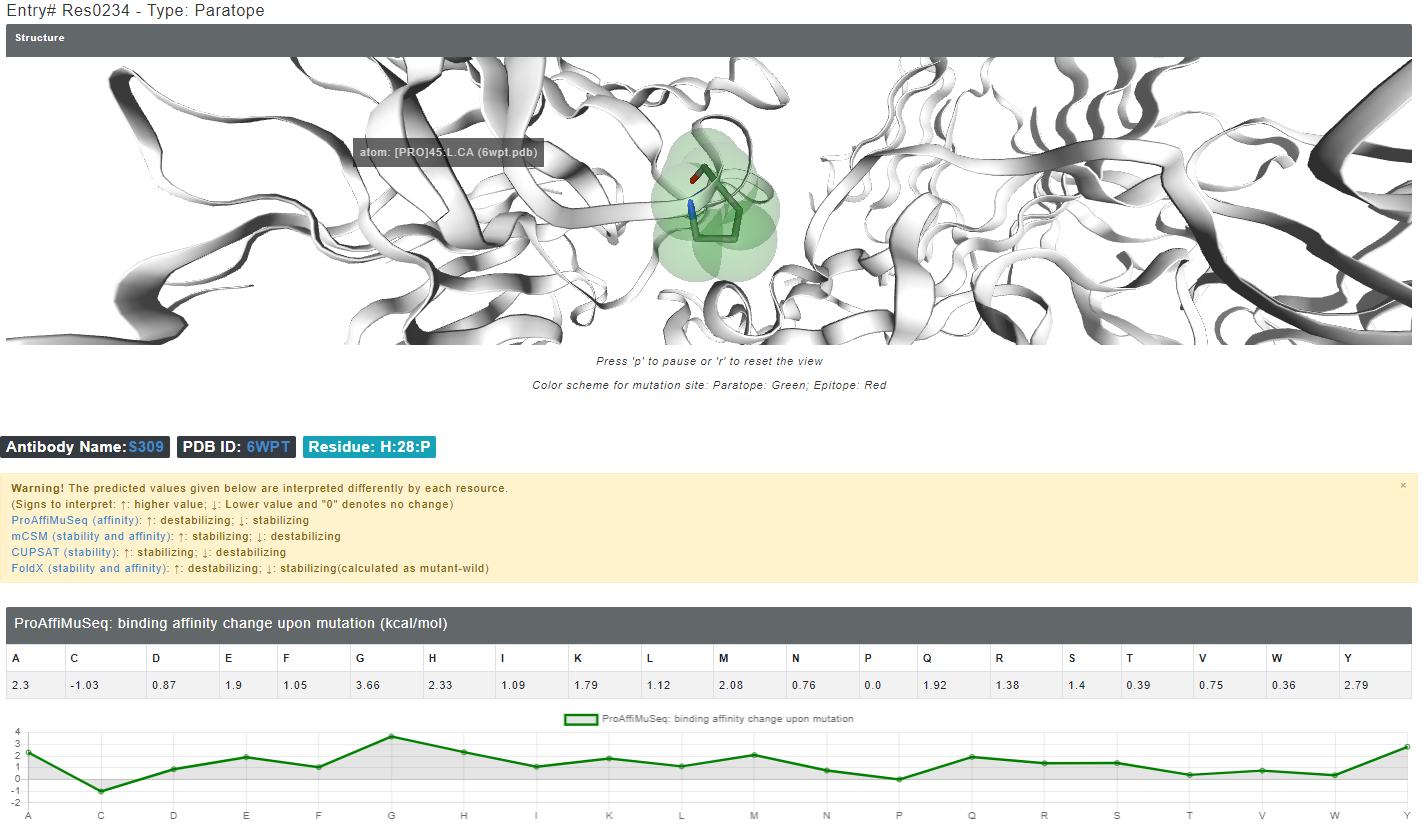
Figure 6. The structure highlights the selected mutation site in green (if paratope) and red (if epitope) colors. Other interface residues can be accessed from the left side table.
A detailed inforamtion is also provided explaining how to interpret the predicted results with links to the original server.
Additional information: dataset of SARS-CoV-2 proteins and related information
Each entry in neutralization profile database also contains viral protein and epitope information. This information is linked with internal dataset of SARS-CoV-2 proteins. We have also calculated features derived from these viral proteins using PDBparam (https://www.iitm.ac.in/bioinfo/pdbparam/index.html). The derived features include contacts (short, medium, long range; CA-CA distance contacts in 8Å and 14Å distance), long range contact order, interactions within protein (ionic, hydrophobic, aromatic-aromatic, cation-pi) and surrounding hydrophobicity
The output page for the viral protein looks like below. The result page displays the structure of the viral protein (experimental structures modelled for missing residues using I-TASSER, taken from https://zhanglab.ccmb.med.umich.edu/COVID-19/), sequence of viral protein with general protein information.
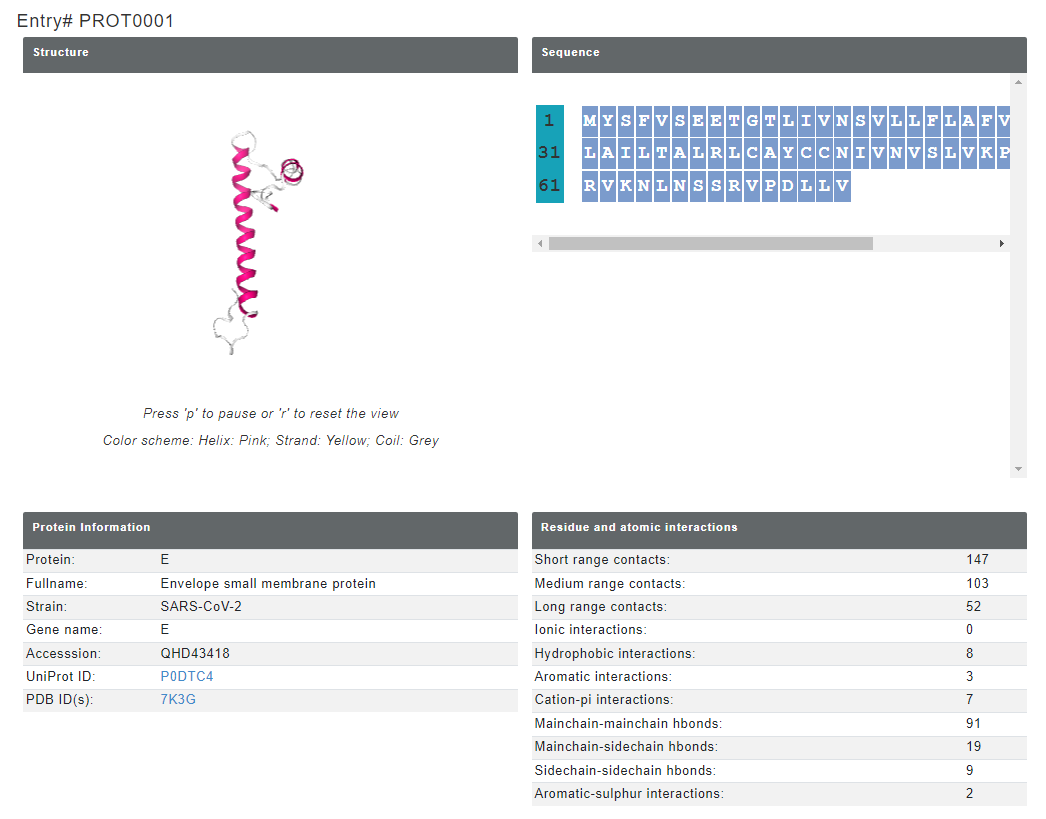
Figure 7. Viral protein information provided for each entry. In the structure, yellow, pink and grey color represents beta sheet, helix and coil respectively.
The precalculated structural features for the viral proteins can be downloaded from the result page. Additionally, this comprehensive information for all proteins is provided with the bulk download option.
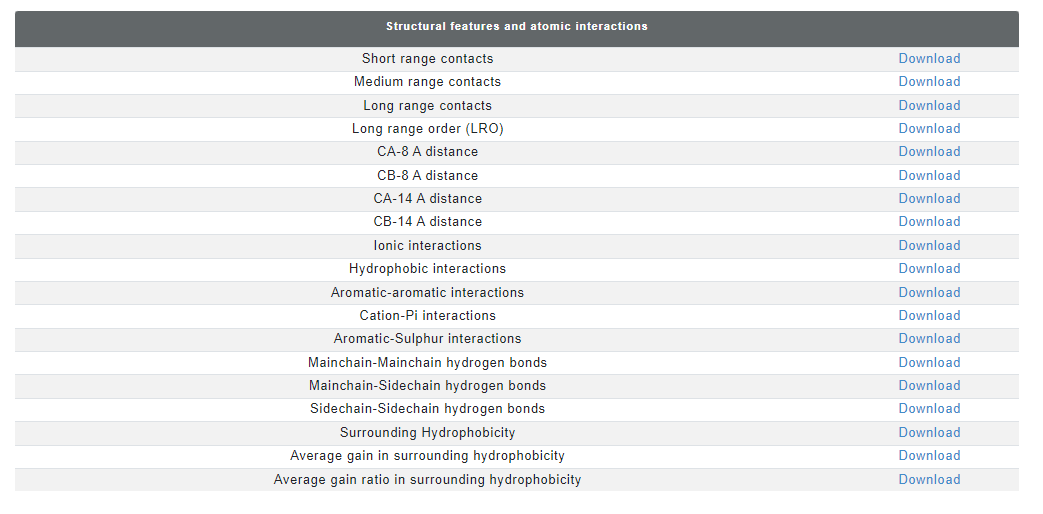
Figure 8. The precalculated features can be downloaded in a CSV file format.
Features calculated for viral proteins
We have calculated several features related to SARS-CoV-2 viral proteins. These features can be useful to analyze the stability, folding dynamics, mutational frequency, biologically relevant residues/regions, drug targets, existing or emerging viral strains etc. These structural features can also be used in the development of machine-learning based models and interpretation of molecular dynamics simulations. Each of these features are discussed below:Short-, medium-, and long-range interactions:
For a given residue, the surrounding residues within a sphere of 8 Å radius are analyzed in terms of their sequence position. Residues within a distance of two residues from the central residue are considered to contribute to short-range interactions, those within a window between three and four residues to medium-range interactions and those more than four residues apart to long-range interactions. Intraprotein contacts has been extensively studied for protein folding and stability.
Long-range order (LRO)
LRO is derived from long-range contacts (contacts between two residues that are close in space and far in the sequence) in the protein structure. In the equation below, i and j are the two contacting residues within a distance of 8 Å, and N represents the total number of residues in the protein. LRO have shown very strong correlation with the folding rate of small proteins.
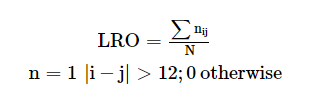
Contacts
The contacts between amino acid residues in the crystal structure are computed with cutoffs of 8 and 14 Å using Cα or Cβ atoms. The residue surrouding any amino acid are useful to understand the local environment, which can be importnat to understand the local stability, flexibility, residue density and mutability. The residue contact features are widely used in the protein structure analysis, ML model development and molecular dynamics studies.
Interresidue interactions
Interactions between amino acid residues include ionic, hydrophobic, aromatic-aromatic, cation-pi, aromatic-sulphur interactions and hydrogen bonds (main chain-main chain, main chain-side chain and side chain-side chain). The interresidue interactions are important for the stability of the proetin structures and are less likely to mutate. The interactions are widely studied in molecular dynamics studies.
Surrounding hydrophobicity
The sum of hydrophobic indices assigned to the residues that appear within a distance of 8 Å from the central residue can be used to characterize the hydrophobic behavior of each amino acid residue in the protein environment. The local exposed hydrophobic patches can be helpful for prediction of functinoal sites in viral proteins including protein binding site(s) and post-translational modification site. These pathces also assist the aggregation of protein. Therefore it can be important feature to study the viral protein or develop ML models. It is defined as:
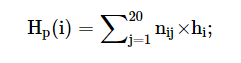
where nij is the total number of surrounding residues of type j around the ith residue of the protein, and hj is the hydrophobicity index (kcal/mol) obtained from thermodynamic transfer experiments. Sometimes surrounding hydrophobicity is not emough to estimate the actual hydrophobicity of the local region. Therefore, an extended unfolded conformation is used as a reference to estimate the gain/gain ratio in hydrophobicity. Gain in surrounding hydrophobicity of a residue is the increase in surrounding hydrophobicity as the protein transitions from its unfolded state to its native (ie, folded) state represents the enrichment in the hydrophobic property of that residue. It is assumed that the fully extended chain conformation is the unfolded reference state and sequence hydrophobicity is calculated for the 2 flanking residues in the sequence. The average gain ratio is the ratio of folded and unfolded surrounding hydrophobicities. It is defined as:
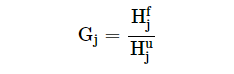
where Hf and Hu denote the hydrophobic index of the jth residue in the folded state and unfolded state of the protein, respectively.
Frequently Asked Questions (FAQ)
Q1. Where can I find the sequences of antibodies in "experimental neutralization profile of coronavirus related antibodies" dataset and therapeutic antibodies?
A1. The sequences and other relevant information for neutralizing antibodies( CoV-AbDab) and therapeutic antibodies (Thera-SAbDab) are present in their respective databases.
Q2. I see therapeutic antibody names in the database, but it is not there in the figshare data.
A2. Therapeutic antibodies are taken from the Thera-SAbDab database (http://opig.stats.ox.ac.uk/webapps/newsabdab/therasabdab/search/). You can download the complete database from there. Similarly, a comprehensive detail of coronavirus related antibodies can be download from CoV-AbDab (http://opig.stats.ox.ac.uk/webapps/covabdab/). Ab-CoV only focuses on experimentally varified neutralizing antibodies.
Q3. Can anyone download the data in bulk?
A3. Yes, anyone can download the bulk data from the figshare repository.
Q4. How to see the computationally predicted affinity and stability for mutations at antibody-spike interface?
A4. There are 2 ways to see it:
Q5. How do I cite Ab-CoV?
A5. The database is under publication stage. we will soon update the information here.
For now, you can cite the figshare DOI: https://figshare.com/s/6e38ad3c2e130a066d19.
Q6. How to use the search page in the Ab-CoV database?
A5. Here are some of the example search in the Ab-CoV datbaase:
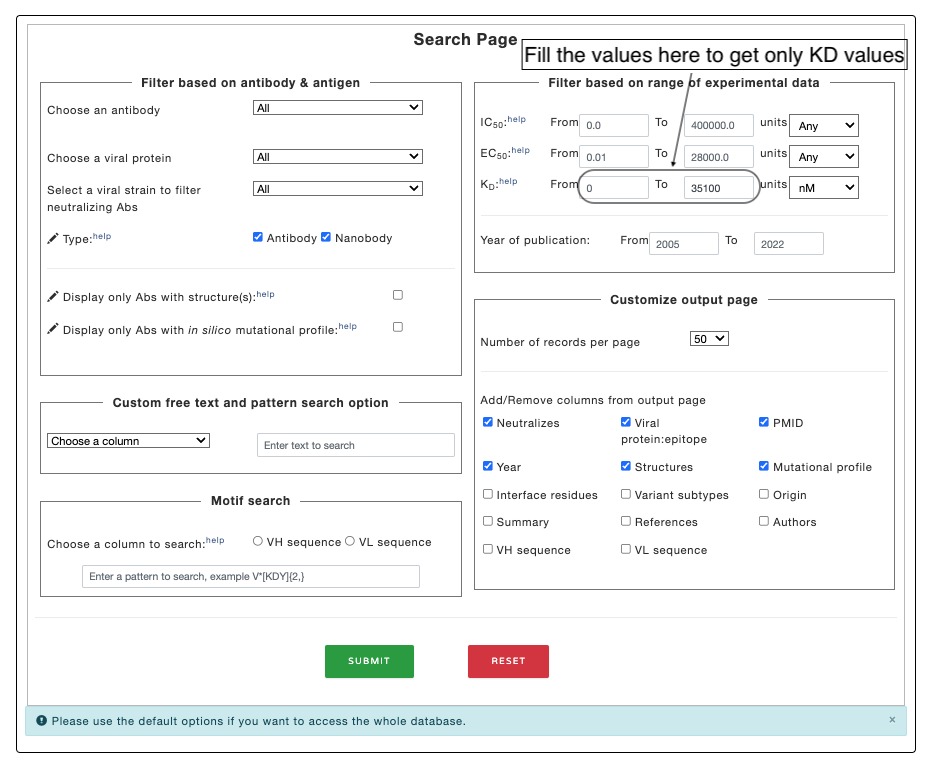
Example 1. Searching only the KD values in the Ab-CoV database.
To search only KD values, Users need to input the range of KD values in the "Filter based on range of experimental data" column in search page. input the prefilled values (lower and upper range) in the respective input boxes to get all the KD values in Ab-CoV database:
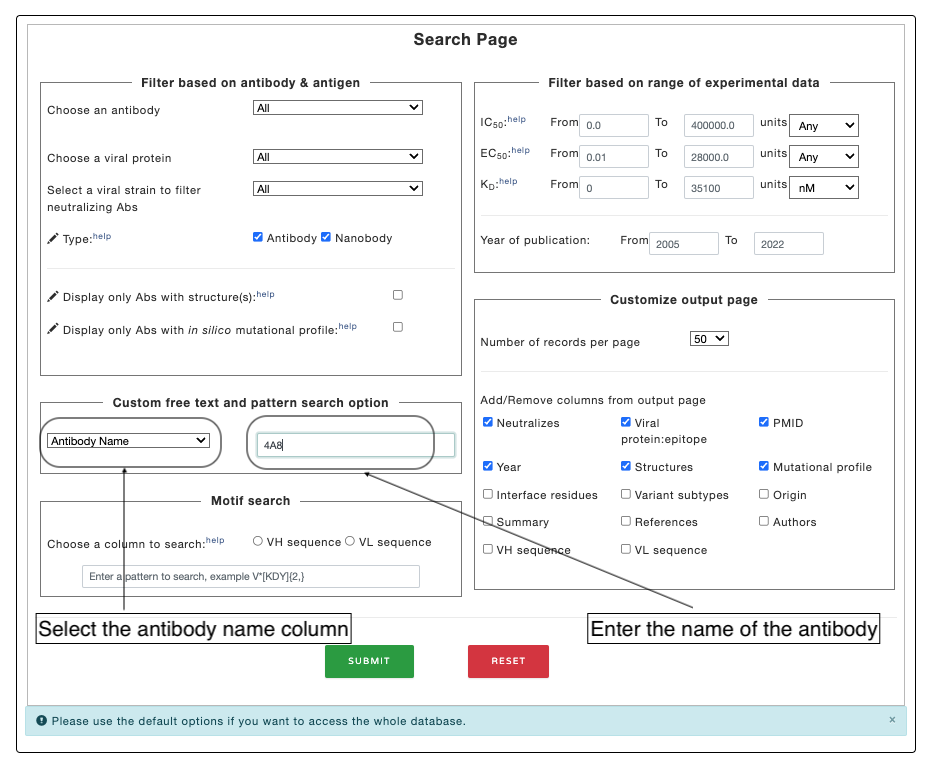
Example 2. Searching specific columns based on values (such as: searching based on antobyd name).
Select the column ("Antiobdy Name" column to search based on name of the antibody) you want to search. Write the text in the adjacent input box and click submit. The database will look of the substring in the respected column.
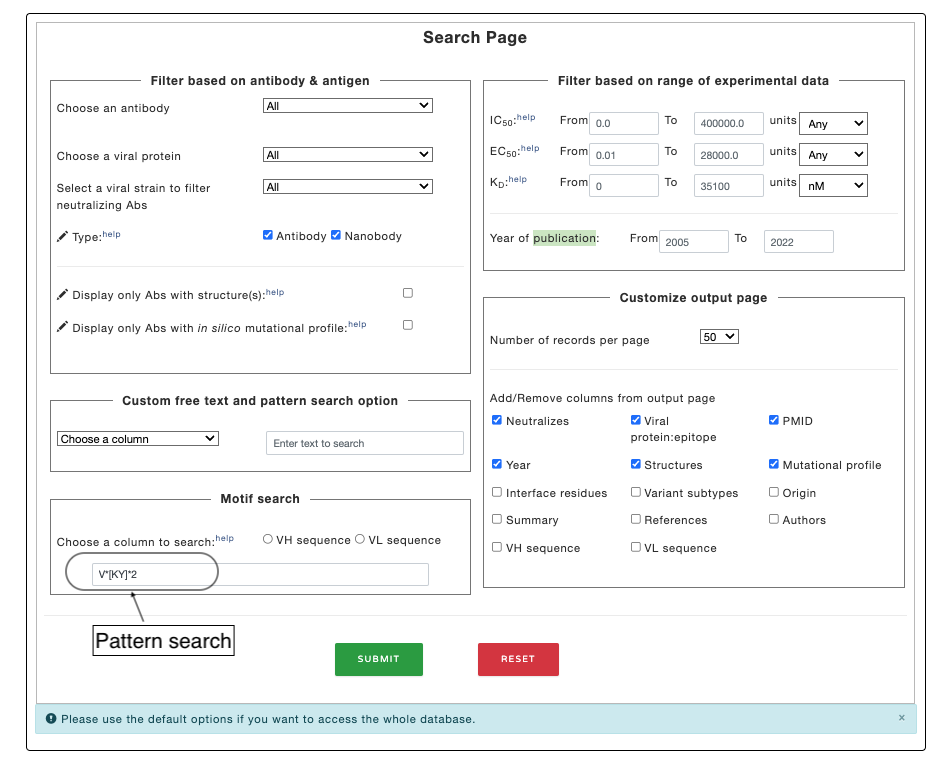
Example 3. Search based on motif.
For more complex search, you can use regular expression to search for specific motif/group of motifs as shown in the figure. Read the wikipedia page for more detail on regular expression. This function is perticularly helpful in searching the antibody sequence for motifs (such as CDR regions).
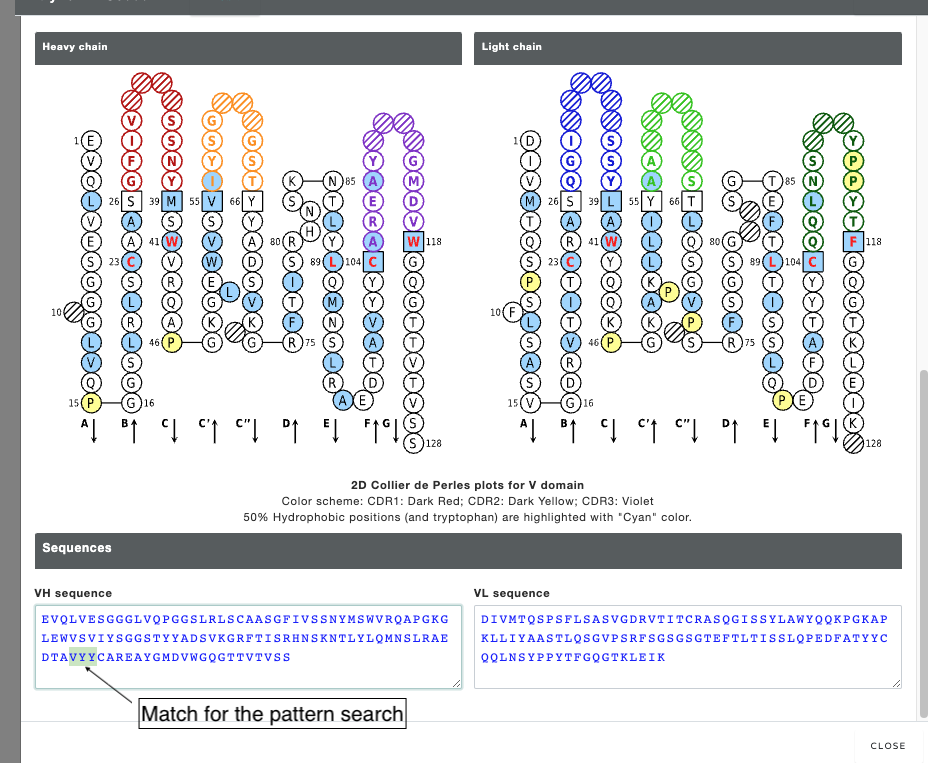
The output of motif search. The searched pattern is highlighted in the image below (Please note that search output does not highlights the output).
About the database:
Ab-CoV database contains information on experimental neutralization assays of coronavirus related antibodies. Additionally, it also provides computationally predicted stability and affinity of mutated paratope/epitope for known spike-antibody complexes, SARS-CoV-2 proteins related information and sequence identity between SARS-CoV-2 neutralizing antibodies and therapeutic antibodies (approved or investigational for other diseases) pairs. These information can be accessed from the home page by clicking the "access the database" button.