A, C, G, U, Purines, Pyrimidines, Total atoms, Total heavy atoms, Total residues, HBA, HBD, Cation, Anion, Polar, Hydrophobic, Others, Molecular surface area, Polar surface area, Non-polar surface area, Relative solvent accessible surface area (SASA), Relative polar SASA, Relative non-polar SASA, Pocket depth, MI1, PMI2, PMI3, NPR1, NPR2, Asphericity, Eccentricity, Spherocity index, Inertial shape factor, R_0.4, R_0.8, R_1.6, R_3.2, alpha, beta, gamma, delta, epsilon, zeta, e_z, chi, phase_angle, ssZp, Dp, splay, eta, theta, eta', theta', eta'', theta'', v0, v1, v2, v3, v4, tm, P, nt_C3'_endo, nt_C2'_endo, nt_C3'_exo, nt_C2'_exo, st_C3’_endo, st_C2’_endo, st_C3’_exo, st_C2’_exo
Overall workflow
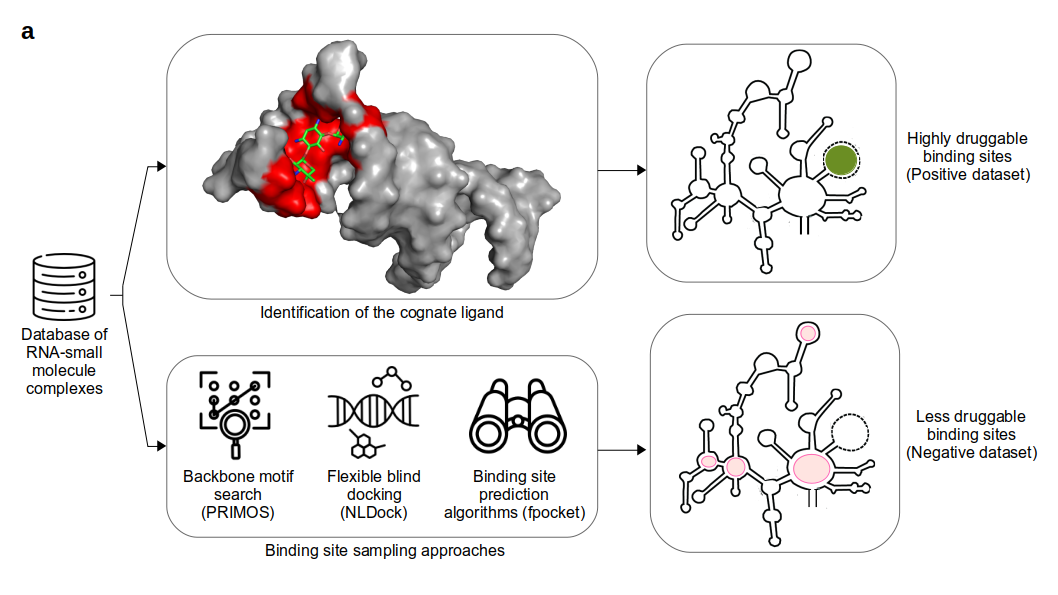
Positive Dataset curation
The dataset used to train the RNA druggability prediction model in DRLiPS server is curated from the Protein Data Bank (PDB) and HARIBOSS databases. A total of 1,073 RNA-small molecule complex structures were collected and pruned to 861 structures to remove structure stabilizing ligands (like spermine), ion-bound riboswitches and synthetic RNA aptamers. This dataset was further sub-divided into 513 ribosomal RNA complexes and 348 non-ribosomal complexes. Out of the 513 ribosomal complexes, only 51 complexes with the aminoacyl tRNA site (A-site) of the 16S rRNA were retained along with non-ribosomal complexes, resulting in a total of 399 structures as our positive dataset (highly druggable binding pockets). Residues within 6.5 Å of the bound ligand were considered to form the RNA-ligand binding pocket.
POSITIVE DATASET STATISTICS
| Total no. of RNA-ligand binding pockets from 399 structures | 1,072 |
| No. of binding pockets after redundancy within structure (homo-multimers) | 819 |
| No. of non-redundant binding pockets after Rfam-based clustering | 56 |
Negative Dataset curation
The negative dataset (less druggable binding pockets) was curated using the following three strategies:
All-to-All flexible blind docking: All unique RNA-binding ligands from PDB were docked to all 399 RNA structures in a blind fashion using the NLDock program. A maximum of 10 binding sites were sampled through docking for each RNA target. All RNA structures were considered to take into account conformational changes in the RNA as captured by experimental methods such as NMR.
Backbone motif search: Backbone-ligand interactions account for less than 10% of ligand selectivity upon RNA binding. Hence, matches in only the backbone motifs with known binding pockets from the positive dataset were considered to be probable negative pockets, since the residue composition of the binding pocket can still differ. The PRIMOS program was used for backbone motif search between unrelated pairs of RNA with a minimum of 4 residues and less than 2.5 Å RMSD as the criteria.
Exhaustive pocket prediction: The fpocket program was used to predict all possible binding sites present in the 399 RNA structures. Redundant binding pockets across multiple representative structures of the same RNA target were merged if they shared 90% of the residues.
Negative binding pockets sampled through all the three above approaches were combined to obtain a dataset of 3947 non-redundant pockets. With a 70% residue overlap cut-off, comparison with positive dataset resulted in 93 negative binding pockets for the final training dataset. In total, 56 positive binding pockets and 93 negative binding pockets were used for the DRLiPS model.
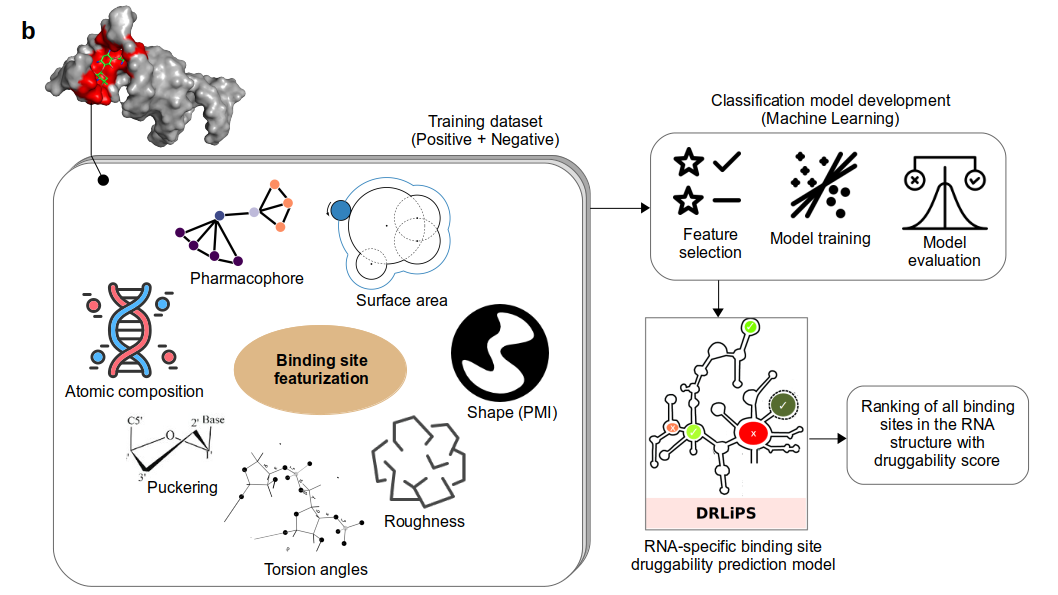
Feature calculations
The features or descriptors to train the classification model were computed using the RNA-ligand binding pockets obtained as described above. Seven classes of binding site features were computed namely: Composition, Pharmacophore, Surface area, Principal Moments of Inertia (Shape), Roughness, Torsions, Nucleotide type and sugar puckering. For a detailed description of the features, please refer to the Methods section of the associated article.
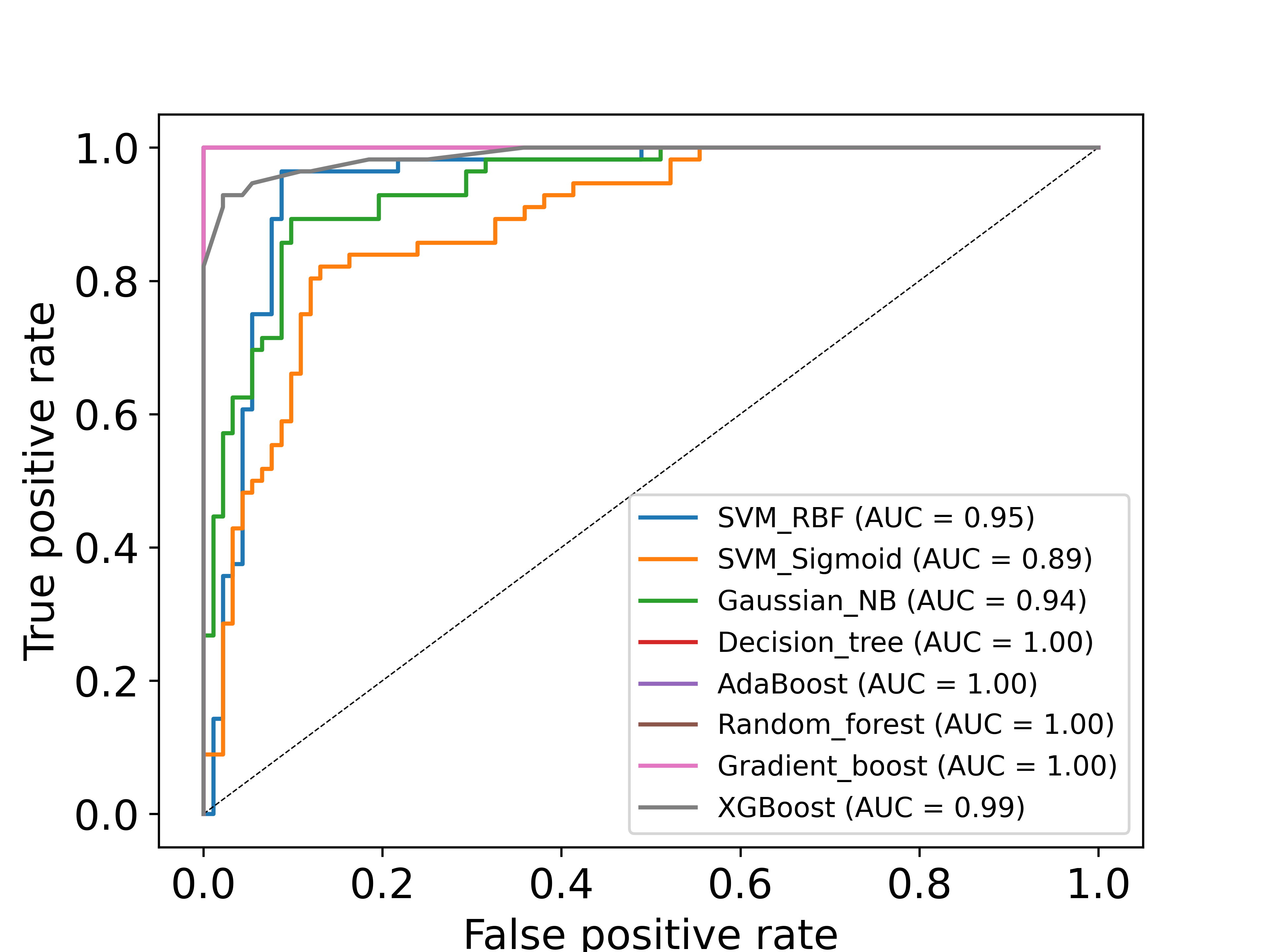
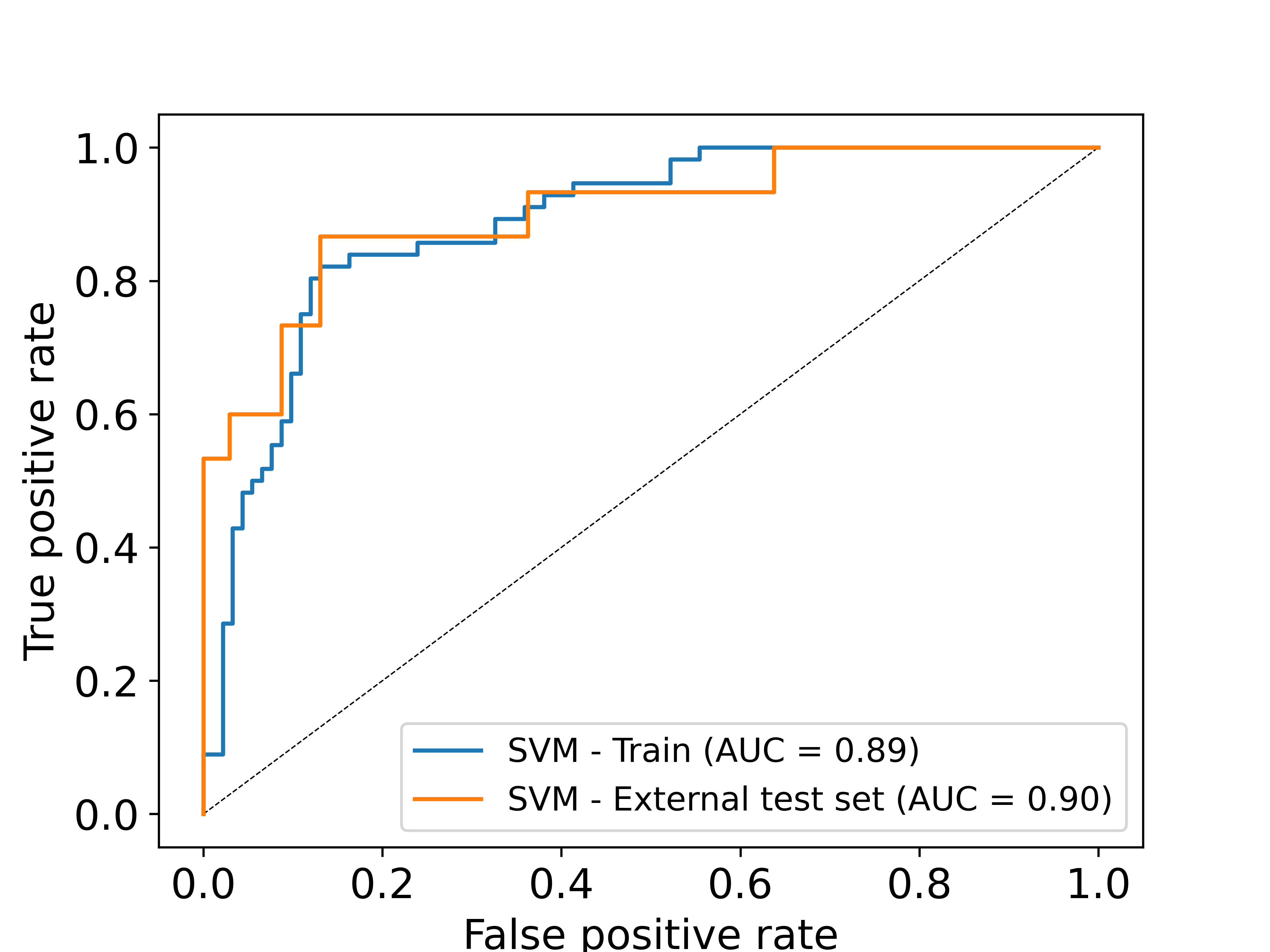
Model performance
The performance of the classification model was quantified using the receiver operating characteristic (ROC) score and F1-score. Multiple machine learning methods were tested out of which, the support vector machine (SVM) model with sigmoid kernel and minmax feature normalization was found to perform the best. For more details on the performance of the models on training set and on external blind test datasets, please refer our associated publication.
Performance plots
More details on how to use DRLiPS for RNA druggability prediction are available in the Tutorial and FAQs page. If you have any further queries on DRLiPS, please contact us.
|
© 2024, Protein Bioinformatics Lab, Indian Institute of Technology Madras All Rights Reserved. This server is maintained by Department of Biotechnology (IIT-M) |