RNA STRUCTURE AS INPUT
The 3D structure of the RNA target of interest must be provided as input in the PDB/ENT format to the prediction form. The model was trained with experimental structures and validated with both experimental and modelled structures. Hence, both experimental and modelled structures are acceptable inputs for prediction.
Examples
| RNA target name | 3D structure of the RNA | PDB format (3D) |
|---|---|---|
| HIV-1 TAR RNA | 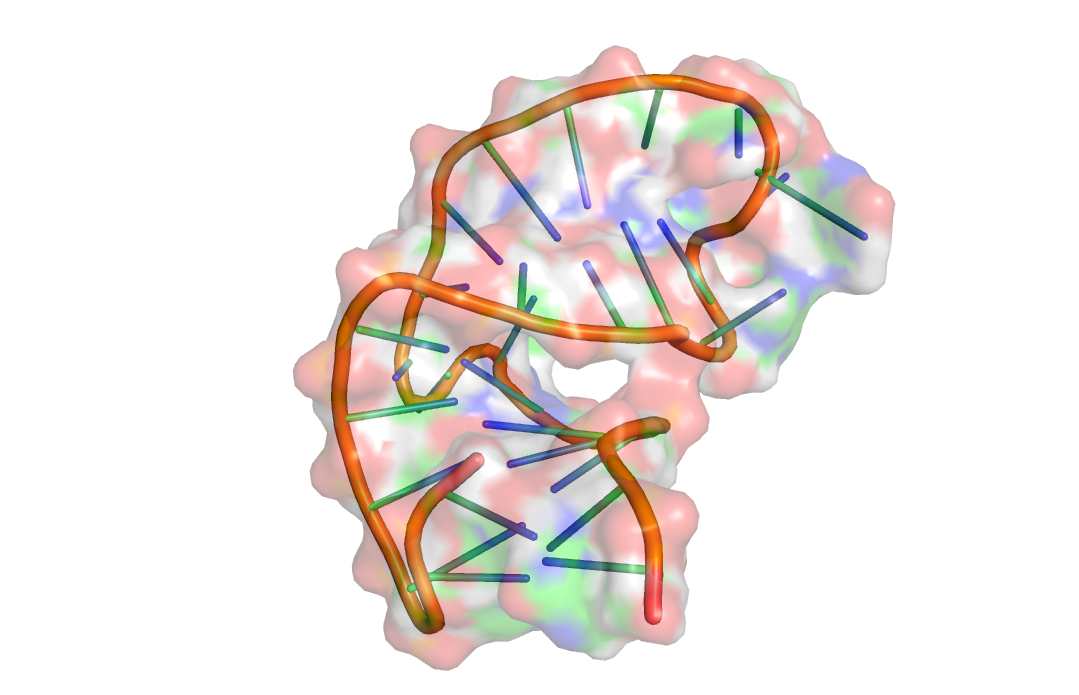 |
|
| FMN riboswitch | 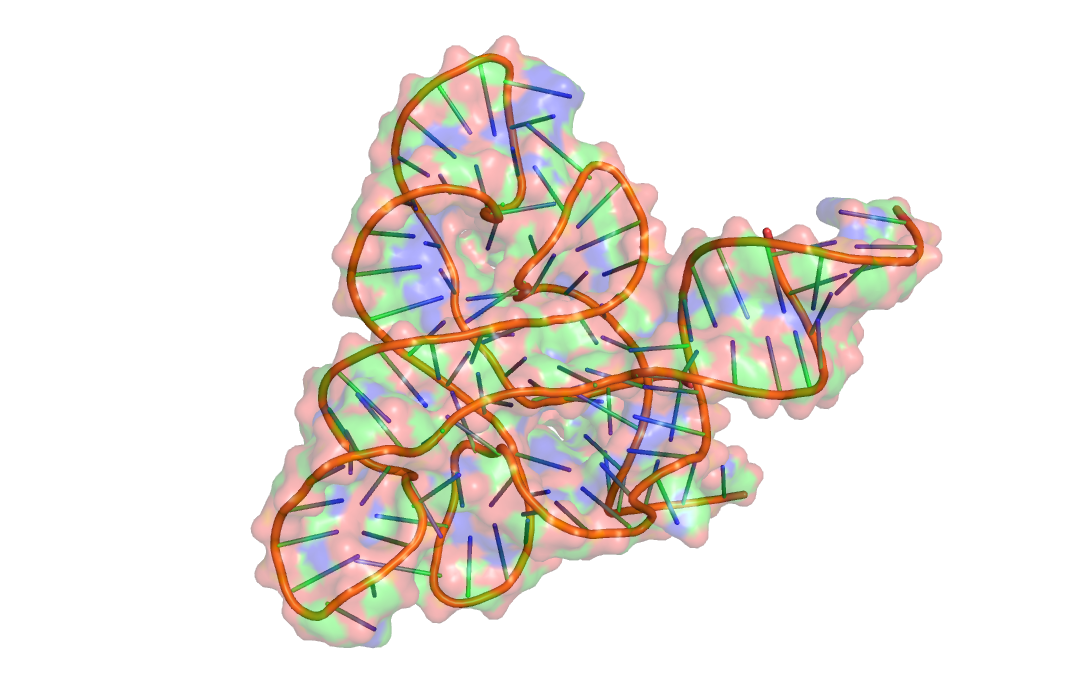 |
|
| Bacterial ribosomal A-site | 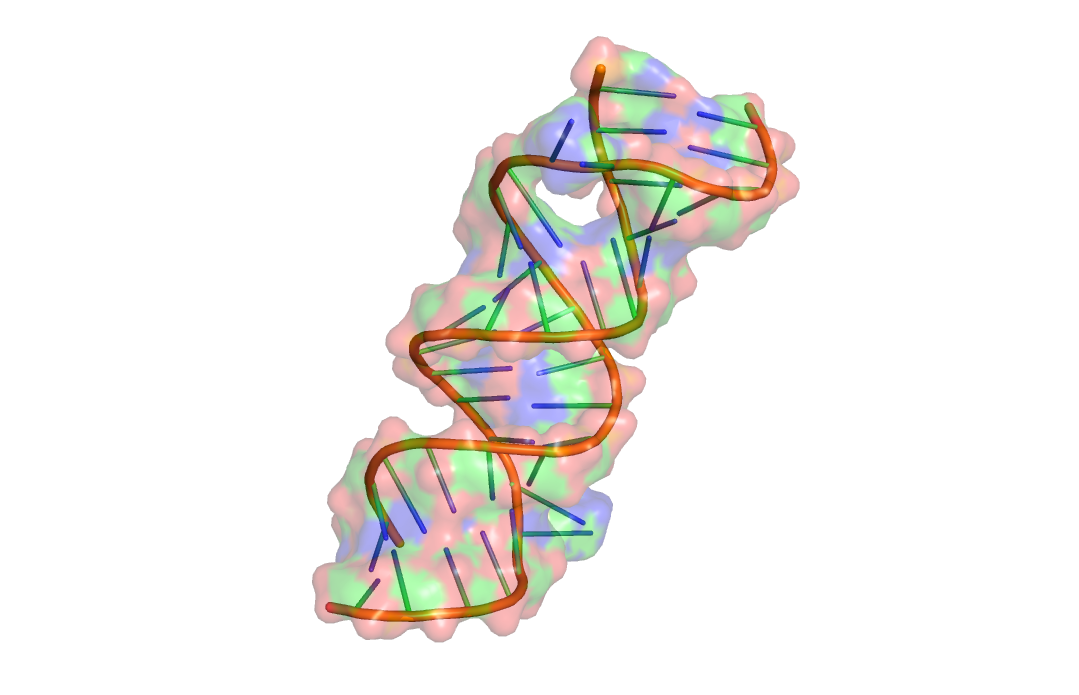 |
REPRESENTATION OF A KNOWN BINDING POCKET
When a binding pocket is known in an RNA target and the user needs to calculate the druggability score for it, the binding pocket residues must be extracted and supplied as input along with the 3D structure of the RNA. Each residue or nucleotide in the binding pocket is defined by a residue identifier with these 3 components: residue one-letter code, residue number in structure, chain ID. These 3 values are joined together using the underscore (_) character to form the residue identifier. All residue identifiers describing the binding pocket can be converted into a comma-separated string, or as a text file with one residue per line and supplied. On the other hand, if the user is aware of the PDB ligand identifier for the bound ligand, it can also be used instead of the binding pocket residues.
Examples
1. Argininamide binding site in HIV-1 TAR RNA: G_21_A, A_22_A, U_23_A, U_25_A, G_26_A, A_27_A, G_28_A, C_29_A, C_30_A, A_35_A, G_36_A, C_37_A, U_38_A, C_39_A
2. CMBL3a binding site in CAG repeats: G_1_A, C_2_A, A_3_A, A_6_A, A_3_B, G_4_B, A_6_B, G_7_B
3. Paramomycin binding site in bacterial A-site: C_3_A, G_4_A, U_5_A, C_6_A, A_7_A, C_8_A, A_9_A
IDENTIFYING BINDING POCKETS
Following the scPDB convention, binding pocket residues are defined based on a 6.5 Å distance cut-off from the heavy atoms of the bound ligand. Any residue/nucleotide in the RNA structure, with at least one atom within the distance range, is considered as part of the binding pocket. In case of in silico pocket predictions using the fpocket program, the residues present in the .pqr output files are considered as the binding pocket residues. A binding pocket with ligand is visualized using Jsmol below for clarity.
3D structure with binding pocket in orange and ligand in sticks
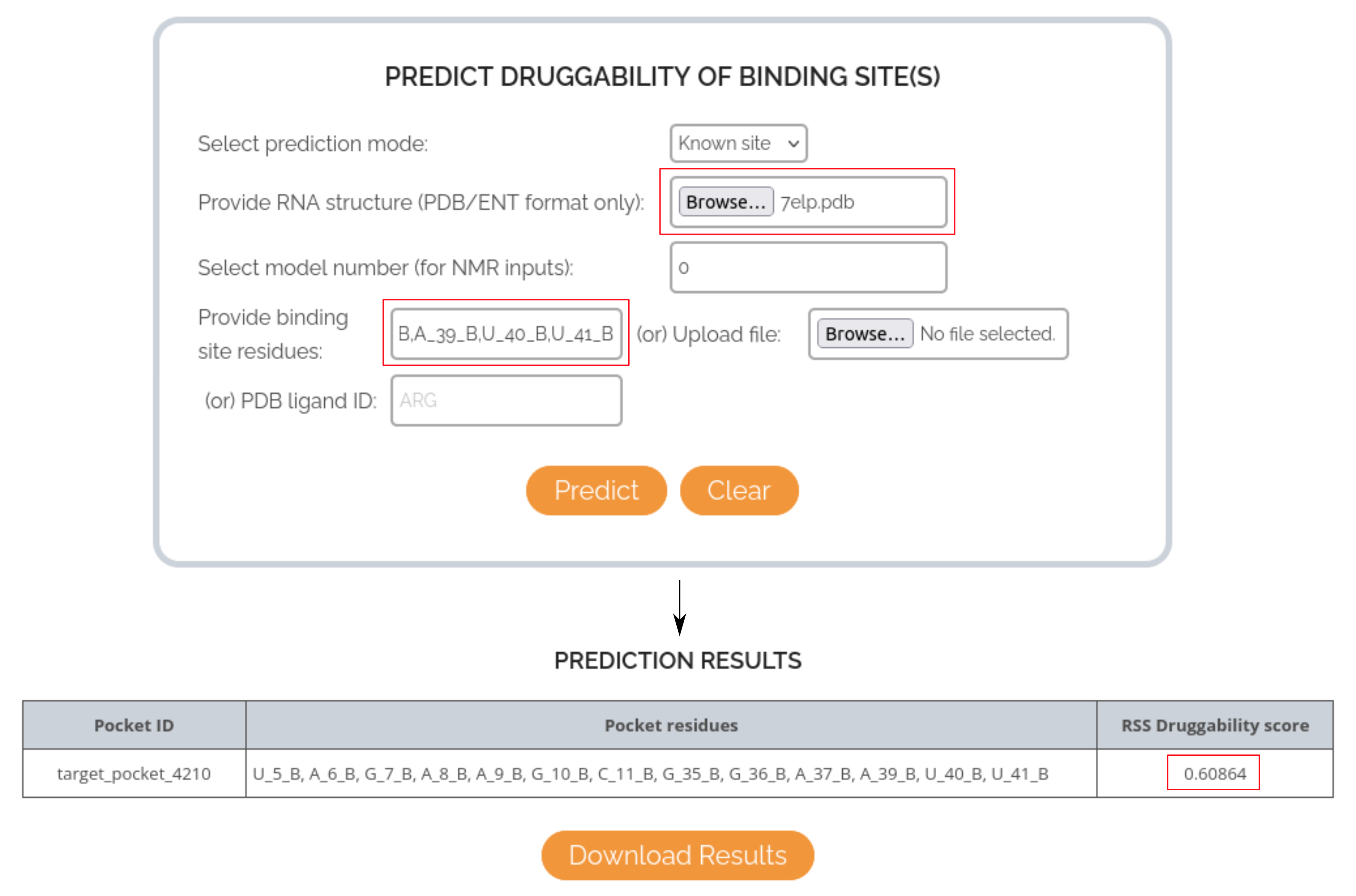
CASE STUDY 1: USING LIGAND IDENTIFIER TO PREDICT THE DRUGGABILITY OF XANTHINE RIBOSWITCH BINDING POCKET
In this case, the RNA target of interest is the Xanthine riboswitch and the small molecule is Xanthine (endogenous substrate). The PDB structure used for this case study is 7ELP and the binding pocket is defined using the ligand ID XAN, as per the input PDB file. The druggability score predicted by RSS for the binding pocket is: 0.60, indicating that the model could correctly predict the druggable nature of the site.
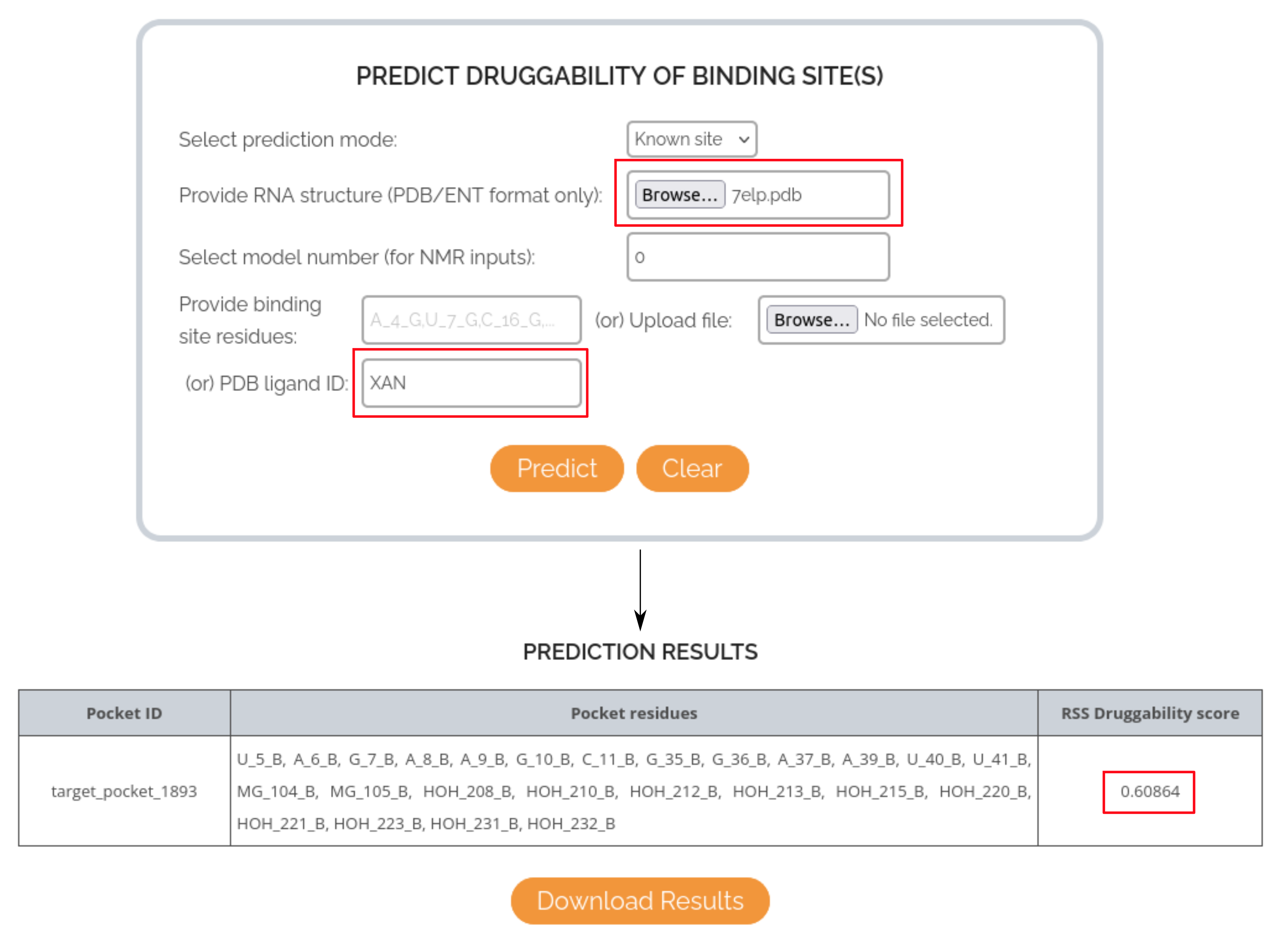
CASE STUDY 2: USING RESIDUE LIST TO PREDICT THE DRUGGABILITY OF XANTHINE RIBOSWITCH BINDING POCKET
In this case, the list of binding site residues (U_5_B, A_6_B, G_7_B, A_8_B, A_9_B, G_10_B, C_11_B, G_35_B, G_36_B, A_37_B, A_39_B, U_40_B, U_41_B) is directly supplied as input to the RSS prediction form, instead of the ligand identifier. The druggability score is still the same as before: 0.60, indicating the deterministic nature of the model prediction, irrespective of the input mode used.
CASE STUDY 3: USING FPOCKET PREDICTIONS TO RANK BINDING POCKETS AS PER THEIR DRUGGABILITY IN XANTHINE RIBOSWITCH
In this case, the binding mode is changed from Known site to All sites, to trigger the usage of fpocket program in the background. Only the input RNA structure has to be supplied and the druggability scores will be predicted for all possible binding pockets in the structure. Users can sort the different pockets based on its RSS score and select the best binding pocket for further investigation. In case of the Xanthine riboswitch, fpocket could identify 17 binding sites in the structure and the scores for each site sorted in descending order are shown below.
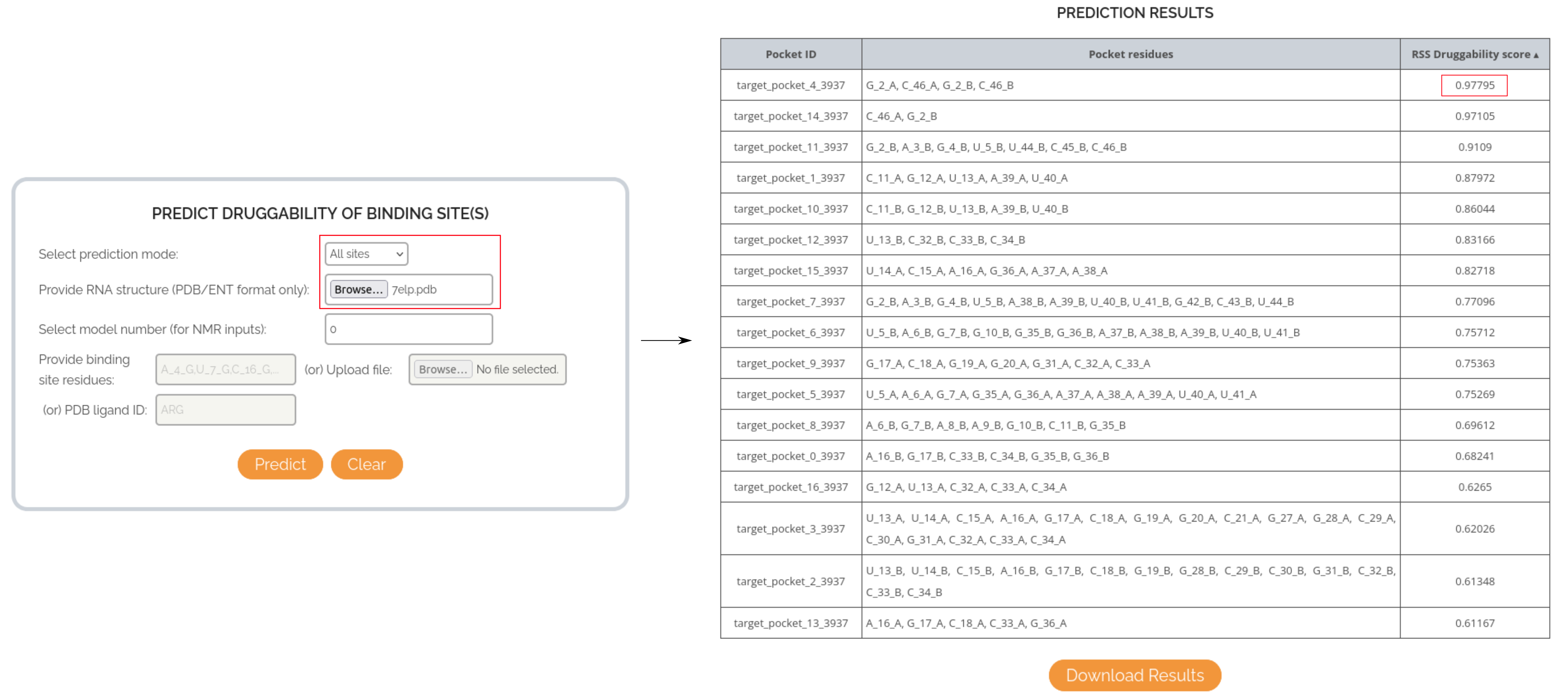
The topmost binding pocket has a score of 0.90, compared to the binding pocket of the endogenous substrate (RSS score = 0.60). This indicates that RSS can help identify potentially highly druggable and unexplored binding pockets within an RNA structure.
|
© 2024, Protein Bioinformatics Lab, Indian Institute of Technology Madras All Rights Reserved. This server is maintained by Department of Biotechnology (IIT-M) |